
UPLC-QTOF-MS测定沉积物中7种新型污染物
2016-02-24 03:27:03谭建华汤嘉骏欧阳培毓王赢利解启来
质谱学报 2016年1期
谭建华,汤嘉骏,欧阳培毓,王赢利,解启来,
阳宇翔1,陈镇新1,刘昕宇1,3
(1.华南农业大学资源环境学院,土壤环境与废物资源农业利用广东高校重点实验室,广东 广州 510642;
2.广州质量监督检测研究院,广东 广州 510110;3.珠江流域水环境监测中心,广东 广州 510611)
UPLC-QTOF-MS测定沉积物中7种新型污染物
谭建华1,2,汤嘉骏1,欧阳培毓1,王赢利1,解启来1,
阳宇翔1,陈镇新1,刘昕宇1,3
(1.华南农业大学资源环境学院,土壤环境与废物资源农业利用广东高校重点实验室,广东 广州510642;
2.广州质量监督检测研究院,广东 广州510110;3.珠江流域水环境监测中心,广东 广州510611)
摘要:本研究建立了固相萃取(SPE)-超高效液相色谱-四极杆飞行时间质谱(UPLC-QTOF-MS)检测沉积物中水杨酸(SA)、萘普生(NAX)、布洛芬(IBU)、酮洛芬(KET)、双氯芬酸(DIC)、降固醇酸(CLO)和三氯生(TRI)7种新型污染物的分析方法。样品用甲酸-丙酮溶液(1∶99,V/V)超声提取,MAX阴离子固相萃取柱净化,UPLC-QTOF-MS测定。在优化的实验条件下,7种新型污染物在2~500 μg/L浓度范围内线性关系良好,线性相关系数均大于0.99,方法检出限(S/N=3)为0.5~0.6 μg/kg,方法定量限(S/N=10)为1.5~2.0 μg/kg(均以干重计),3个添加浓度水平(低、中、高)的回收率为87.8%~105.1%,相对标准偏差(n=5)为3.1%~12.2%。利用该方法对广州市流溪河段表层沉积物样品进行分析,检出4种污染物,最高浓度范围为6.51~12.26 μg/kg(以干重计)。该方法高效、准确,适用于沉积物中7种污染物的测定。
关键词:超高效液相色谱-四极杆飞行时间质谱(UPLC-QTOF-MS);沉积物;新型污染物;固相萃取(SPE)
doi:10.7538/zpxb.2016.37.01.0052
近年来,环境中的新型微量污染物,如药品、个人护理品、内分泌干扰物以及各种工业添加剂等在国际上引起了广泛关注[1-4]。新型污染物在环境中的分布主要通过水相传递和食物链扩散,进而可能对生态系统和人类健康产生危害[5-6]。因此,建立快速、可靠、灵敏的定性定量分析方法,研究新型污染物在各种水环境(河流、海洋、地下水、沉积物、水生动植物)中的浓度水平、分布特征和迁移转化状况,对了解此类物质的污染现状及可能造成的生态影响具有十分重要的意义。
目前,对环境中新型污染物的检测对象主要为各种水体[7-10],沉积物[11]和土壤[12-13]等环境介质。检测方法主要有气相色谱-质谱法(GC/MS)、高效液相色谱-质谱法(HPLC/MS)、液相色谱-串联质谱法(LC-MS/MS)[14-17]等。虽然这些方法的灵敏度和特异性较好,但在定性准确度方面尚有欠缺,容易产生假阳性结果[18]。而四极杆-飞行时间质谱(Q-TOF)技术可对化合物进行精确分子质量测定,并依据所测得化合物的精确分子质量对其进行确证分析,准确度较高。
本工作参考相关文献[19-21],选择在我国环境中暴露水平较高的7种药物和抗菌剂,包括水杨酸、双氯芬酸、酮洛芬、降固醇酸、萘普生、布洛芬和三氯生等,根据其结构性质,结合沉积物的基质特点,通过优化提取、净化的前处理方法,拟建立阴离子交换固相萃取柱净化、超高效液相色谱-四极杆飞行时间质谱(UPLC-QTOF-MS)测定沉积物中上述7种新型微量污染物的检测方法,并将其应用于实际样品的分析。
1实验部分
1.1仪器与试剂
超高效液相色谱-四极杆串联飞行时间质谱仪:美国Waters公司产品;FD-1B-80冷冻干燥机:北京博医康实验仪器有限公司产品;Beckman高速冷冻离心机(Allegra 64R),MS3 basic漩涡混合器:德国IKA公司产品;BG-02C型超声波发生器:广州邦洁超声设备有限公司产品;24孔位固相萃取装置:上海安谱有限公司产品;Milli-Q超纯水系统:美国Millipore公司产品;PVDF滤膜(直径25 mm,孔径0.2 μm):美国Agilent公司产品;HLB固相萃取柱(20 g/L),MAX固相萃取柱(20 g/L):美国Waters公司产品;氮吹仪:美国JNC公司产品。
水杨酸(salicylic acid, SA)、萘普生(naproxen, NAX)、布洛芬(ibuprofen, IBU)、酮洛芬(ketoprofen, KET)、双氯芬酸(diclofenac, DIC)、降固醇酸(clofibric acid, CLO)、三氯生(triclosan, TRI)7种标准品:纯度均大于99.0%,德国Dr. Ehrenstorfer公司产品;甲醇、丙酮:均为HPLC级,美国Fisher 公司产品;甲酸、氨水:均为分析纯,上海安谱公司产品。
1.2样品采集与保存
样品:采集广州市流溪河表层沉积物,用锡箔纸包裹,并于当天运回实验室。样品于-40 ℃真空冷冻干燥48 h,经研钵研磨后,过80目筛,密封避光保存于-20 ℃冰箱中,备用。
1.3样品前处理
1.3.1样品提取准确称取1 g(精确至0.001 g)沉积物样品于10 mL具塞比色管中,加入5 mL含1%甲酸的丙酮溶液,于漩涡振荡器上混合,使样品分散1 min,超声提取5 min,取上清液于离心管中,以10 000 r/min离心5 min,转移至10 mL具塞比色管中。分别用3 mL 和2 mL甲酸-丙酮溶液各提取残渣1次,合并上清液,于35 ℃水浴中氮吹至近干,用1 mL甲醇超声助溶后,加入1 mL超纯水稀释,再加入100 μL氨水,混匀,待固相萃取净化。
1.3.2样品净化用3 mL甲醇、3 mL超纯水活化平衡MAX固相萃取柱,样品溶液以不高于1 mL/min流速通过萃取小柱后,依次用3 mL甲醇淋洗,5 mL甲酸-甲醇溶液(1∶9,V/V)洗脱,收集洗脱液,于35 ℃水浴中氮吹至近干,加1 mL甲醇复溶,过滤,待测。
1.4实验条件
1.4.1色谱条件色谱柱:HSS T3柱(2.1 mm×100 mm×1.8 μm);柱温:30 ℃;进样体积:5 μL;流速:0.3 mL/min;流动相:A为0.002%甲酸水溶液,B为甲醇;梯度洗脱程序:0~1.5 min、40%~65%B,1.5~5.5 min、 65%~100%B,5.5~6.5 min、100%B,6.5~8 min、100%~40%B。
1.4.2质谱条件离子源:电喷雾离子源(ESI),负离子模式;毛细管电压:2.5 kV;萃取锥孔电压:12 V;脱溶剂气温度:400 ℃;离子源温度:100 ℃;脱溶剂气流速:600 L/h;锥孔气流速:50 L/h;四极杆采集质量数范围:m/z80~1 000;数据采集模式:棒状(centroid);扫描采集时间:0.2 s;TOF运行模式:V模式;以200 μg/L亮氨酸脑啡肽(m/z554.261 5)溶液进行实时校准。
2结果与讨论
2.1色谱条件的优化
流动相的组成会影响目标化合物的色谱峰形和离子化效率。由于7种目标化合物均含有羧基或酚羟基结构,本实验选择甲醇和适量浓度的甲酸水溶液为流动相。在该流动相体系中,目标化合物均能得到较理想的峰形。但进一步实验发现,水相流动相中高浓度的甲酸会对目标化合物产生离子抑制效应。因此,实验考察了水相流动相中甲酸浓度为0.002%~0.05%时对目标化合物的影响。结果表明,流动相中甲酸浓度在0.002%时,目标化合物的离子抑制效应不明显,能保证较好的色谱峰形;但随着甲酸浓度的增加,离子抑制效应不断增强。综合考虑流动相的稳定性和方法的灵敏度,最终采用甲醇-0.002%甲酸水溶液作为流动相。
2.2质谱条件的优化
实验对毛细管电压、锥孔电压、脱溶剂气温度、离子源温度、脱溶剂气流速等质谱条件进行了优化。结果发现,7种目标化合物的分子离子不稳定,可能存在源内裂解现象,在未施加碰撞电压的条件下即能获得丰度较高的碎片离子。因此,为了获得最佳灵敏度,采用在全扫描模式下进行选择离子监测,即同时提取目标物的准分子离子和1个碎片离子,并根据出峰时间和离子丰度比进行定性筛查。对于阳性样品,则需选择母离子(准分子离子)进行二级质谱扫描,通过碎片离子定性确证。另外,在优化的质谱条件下,选择响应丰度最高的离子进行定量分析。定量时,使用宽度为0.05 Da的窗口,输入各化合物的精确离子质量数,得到相应的提取离子色谱图,示于图1。
通过MassLynx软件中Elemental Composition分析,得到目标化合物的元素组成与对应理论精确分子质量相匹配的质量精确度。在实际实验中,质量精确度绝对值小于5×10-6即可实现对目标化合物的准确定性。各化合物的质谱参数、保留时间、精确分子质量、理论分子质量和质量精确度列于表1。7种目标化合物的质量精确度绝对值在0.4×10-6~4.7×10-6之间。根据欧盟2002/657/EC的规定,每个高分辨离子可以得到2个确证分(IPS)。因此,本实验的定性筛查和定性确证方法均具有较高的准确性。

注a:双氯芬酸;b.酮洛芬;c.萘普生;d.降固醇酸;e.三氯生;f.布洛芬;g.水杨酸图1 7种目标化合物标准溶液(100 μg/L)的提取离子流色谱图Fig.1 Representative extracted ion chromatograms of seven target analytes in standard solution (100 μg/L)

化合物保留时间/min元素组成锥孔电压/V精确分子质量/Da理论分子质量/Da质量误差/mDa/10-6双氯芬酸4.62C14H10NO2Cl212294.0087*294.0089-0.2-0.7C13H10NCl2250.0191250.01900.10.4酮洛芬3.54C16H13O312253.0866253.08650.10.4C15H13O209.0965*209.0966-0.1-0.5三氯生5.23C12H6O2Cl312286.9427*286.9433-0.6-2.1C12H6O2Cl3288.9410288.94050.51.7萘普生3.73C14H13O312229.0859229.0865-0.6-2.6C12H10O170.0730*170.0732-0.21.1降固醇酸3.82C10H10O3Cl12213.0316*213.0318-0.2-0.9C6H4OCl126.9957126.99510.64.7布洛芬4.72C13H17O212205.1235*205.12290.62.9C12H17161.1329161.1330-0.10.6水杨酸2.37C7H5O312137.0237*137.0238-0.1-0.7C6H5O93.034493.03400.44.3
注:*表示定量离子
2.3样品前处理条件的优化
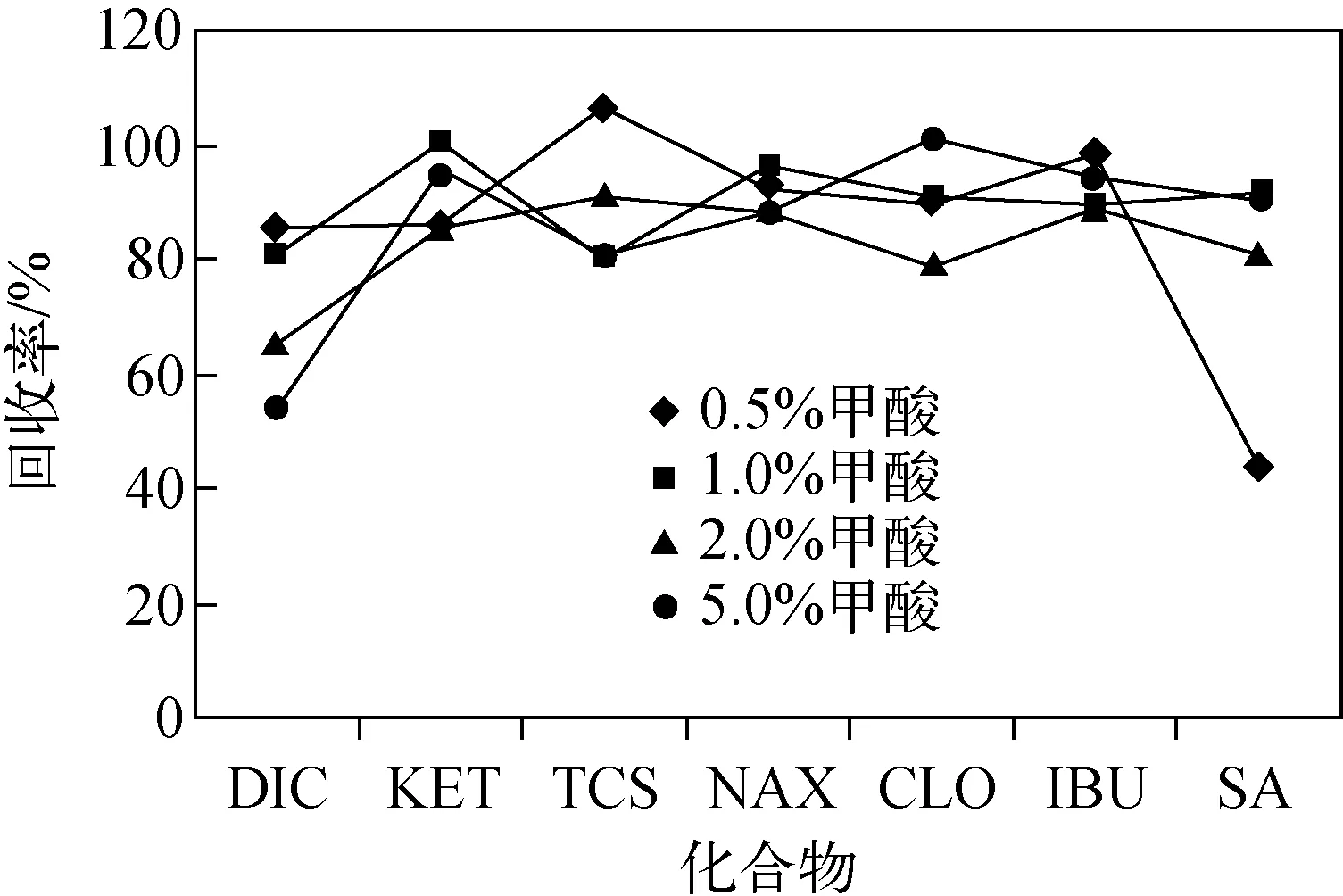
图2 甲醇提取液中加入不同浓度甲酸时的回收率Fig.2 Recoveries obtained with methanolsolutions containing formic acidat different concentrations
2.3.1提取方法的优化通过比较甲醇、丙酮、乙酸乙酯对沉积物样品的提取效果,发现3种溶剂对TRI以外的6种目标化合物的提取效率较差,回收率均低于60%。已有研究表明,目标化合物在沉积物中的提取效率与溶剂的pH值有关[22],在溶剂中加入一定比例的酸能够明显改善提取效率[23-24]。因此,本实验考察了在甲醇溶剂中加入不同体积浓度(0.5%、1.0%、2.0%、5.0%)的甲酸对目标化合物提取效率的影响,结果示于图2。图2显示,加入甲酸能够明显改善对大部分目标化合物的提取效率,在甲酸添加量为1%时,各化合物的回收率均在80%以上。但随着甲酸浓度的升高,DIC的回收率呈下降趋势,这可能与其在沉积物中的吸附性有关。DIC含有1个仲胺结构,由于与相邻的2个苯环共轭,导致N原子上的孤对电子部分转向苯环,碱性非常弱。因此,在甲酸含量较低的提取液中,N原子很难被离子化形成铵盐,但是,随着提取液中甲酸含量的增加,部分DIC可能慢慢地被离子化,从而与沉积物中的腐殖酸等有机物发生强吸附作用而导致提取效率降低。另外,实验还比较了1%甲酸-甲醇溶液,1%甲酸-丙酮溶液,1%甲酸-乙酸乙酯溶液对目标化合物的提取效果,结果示于图3。可见,1%甲酸-丙酮溶液对目标化合物的提取效率最高,回收率均在85%以上。因此,本实验选择1%甲酸-丙酮溶液作为提取溶剂。

图3 不同提取剂的提取效果Fig.3 Recoveries obtainedwith different extraction solutions
2.3.2固相萃取方法的优化固相萃取柱的选择除考虑对目标化合物的富集作用,还要考虑对杂质的去除作用。本实验比较了两种不同填料类型的固相萃取小柱(HLB、MAX)的净化效果。由于沉积物环境基质复杂,有机质含量较高,提取液呈黄色或黄褐色[25-26],以反相保留为主的HLB小柱去除基质干扰效果较差。而MAX小柱为聚合物基质的混合型强阴离子交换柱,同时具有阴离子交换和反相保留能力,7种目标化合物在碱性条件下离子化,与MAX柱上的强阴离子交换树脂进行吸附,通过甲醇淋洗萃取柱,能将具有反相保留的中性、碱性杂质去除。综上考虑,本实验选择MAX小柱为沉积物样品的萃取净化柱。

图4 不同洗脱溶剂的回收率Fig.4 Recoveries obtained with different elution solutions
实验还对固相萃取的洗脱条件进行了优化。分别比较了含5%、10%、15%、20%甲酸的甲醇溶液为洗脱溶剂的洗脱效果,结果示于图4。结果表明,由于SA具有较强的酸性,5 mL含5%甲酸的甲醇溶液对SA无法完全洗脱,即使洗脱体积增加到10 mL,其回收率仍低于50%;而在甲酸含量高于10%的洗脱剂条件下,7种目标化合物的回收率均高于90%。因此,本实验选择5 mL含10%甲酸的甲醇溶液为洗脱溶剂。
2.4线性范围和方法检出限
配制浓度为2.0~500 μg/L的系列标准溶液,在优化好的实验条件下测试,以提取目标离子峰面积y对相应的质量浓度x(μg/L)进行线性回归,并绘制标准曲线。以定量离子3倍信噪比(S/N=3)的响应值对应的化合物浓度为方法检出限(LOD),以10倍信噪比(S/N=10)为方法定量限(LOQ)。7种目标化合物的线性方程、相关系数、检出限和定量限列于表2。由表2可见,7种目标化合物在2.0~500 μg/L范围内具有良好的线性关系(R2>0.99),检出限为0.5~0.6 μg/kg,定量限为1.5~2.0 μg/kg。

表2 目标化合物的线性方程、线性范围、相关系数、方法检出限(LOD)和定量限(LOQ)
2.5方法回收率和精密度
向沉积物样品中分别添加不同浓度水平(2、10和20 μg/kg)的混合标准溶液,平行测定5次,计算回收率和精密度,结果列于表3。结果表明,各化合物的平均回收率在87.8%~105.1%之间,相对标准偏差为3.1%~12.2%,能够满足环境样品中痕量污染物分析的要求。

表3 回收率和精密度测定结果(n=5)
2.6实际样品的分析
在广州市流溪河全河段采集11个表层沉积物样品,利用本方法进行7种污染物的分析测定。结果显示,从沉积物样品中检出SA、TRI、IBU和KET 4种化合物,其浓度范围分别在 3结论 采用阴离子交换固相萃取柱净化技术,结合超高效液相色谱-四极杆飞行时间质谱建立了沉积物中7种新型微量污染物的检测方法。通过对提取条件、固相萃取条件、色谱及质谱参数的优化,有效地提高了方法的灵敏度和准确性。采用选择离子监测模式,结合离子的精确质量数、同位素丰度比、离子相对丰度和出峰时间进行定性筛查分析;阳性样品通过对母离子(准分子离子)二级质谱扫描,采集碎片离子进行定性确证。结果表明,本方法具有较强的确证性,大大降低了假阳性结果的产生,能够满足复杂沉积物基体中7种新型微量污染物测定的要求。 参考文献: [1]MOLDOVAN Z. Occurrences of pharmaceutical and personal care products as micropollutants in rivers from Romania[J]. Chemosphere, 2006, 64(11): 1 808-1 817. [2]ELLIS J B. Pharmaceutical and personal care products (PPCPs) in urban receiving waters[J]. Environ Pollut, 2006, 144(1): 184-189. [3]WU M, JANSSEN S. Dosed without prescription: A framework for preventing pharmaceutical contamination of our nation’s drinking water[J]. Environ Sci Technol, 2011, 45(2): 366-367. [4]VALCARCEL Y, GONZALEZ ALONSO S, RODRIGUEZ-GIL J L, et al. Analysis of the presence of cardiovascular and analgesic/anti-inflammatory/antipyretic pharmaceuticals in river- and drinking-water of the Madrid Region in Spain[J]. Chemosphere, 2011, 82(7): 1 062-1 071. [5]ZHOU X F, DAI C M, ZHANG Y L, et al. A preliminary study on the occurrence and behavior of carbamazepine (CBZ) in aquatic environment of Yangtze River Delta, China[J]. Environ Monit Assess, 2011, 173(1): 45-53. [6]ZORITA S, MARTENSSON L, MATHIASSON L. Trace element deposition and trends during a ten year period in Finland[J]. Sci Total Environ, 2009, 407(7): 2 260-2 270. [7]LEE H B, PEART T E, SVOBODA M L. Determination of endocrine-disrupting phenols, acidic pharmaceuticals, and personal-care products in sewage by solid-phase extraction and gas chromatography-mass spectrometry[J]. J Chromatogr A, 2005, 1 094(1/2): 122-129. [8]贾妍艳,谭建华,徐晨,等. 固相萃取-气相色谱-质谱法同时测定水中9种药品及个人护理用品[J]. 色谱,2014,32(3):263-267. JIA Yanyan, TAN Jianhua, XU Chen, et al. Simultaneous determination of nine pharmaceuticals and personal care products in waters by solid phase extraction-gas chromatography-mass spectrometry[J]. Chinese J Chromatography, 2014, 32(3): 263-267(in Chinese). [9]TARCOMNICU I, ALEXANDER L N, NUIJS V, et al. Simultaneous determination of 15 top-prescribed pharmaceuticals and their metabolites in influent wastewater by reversed-phase liquid chromatography coupled to tandem mass spectrometry[J]. Talanta, 2011, 83(3): 795-803. [10]BUCHBERGER W W. Current approaches to trace analysis of pharmaceuticals and personal care products in the environment[J]. J Chromatogr A, 2011, 1 218(4): 603-618. [11]MINTEN J, ADOLFSSON-ERICI M, ALSBERG T. Extraction and analysis of pharmaceuticals in polluted sediment using liquid chromatography mass spectrometry[J]. Int J Environ Anal Chem, 2011, 91(6): 553-566. [12]RICE S L, MITRA S. Microwave-assisted solvent extraction of solid matrices and subsequent detection of pharmaceuticals and personal care products (PPCPs) using gas chromatography-mass spectrometry[J]. Anal Chim Acta, 2007, 589(1): 125-132. [14]DUAN Y P, DAI C M, ZHANG Y L, et al. Selective trace enrichment of acidic pharmaceuticals in real water and sediment samples based on solid-phase extraction using multi-templates molecularly imprinted polymers[J]. Analytica Chimica Acta, 2013, 758(1): 93-100. [15]YONG Y, WU L. Analysis of endocrine disrupting compounds, pharmaceuticals and personal care products in sewage sludge by gas chromatography-mass spectrometry[J]. Talanta, 2012, 89(2): 258-63. [16]VERGA M, DOBOR J, HELENKR A, et al. Investigation of acidic pharmaceuticals in river water and sediment by microwave-assisted extraction and gas chromatography-mass spectrometry[J]. Microchem J, 2010, 95(2): 353-358. [17]HILTON M J, THOMAS K V. Determination of selected human pharmaceutical compounds in effluent and surface water samples by high-performance liquid chromatography-electrospray tandem mass spectrometry[J]. J Chromatogr A, 2003, 1 015(1/2): 129-141. [18]张峰,许成保,蓝芳,等. 超高效液相色谱-四极杆飞行时间质谱法测定饲料中9种雄性激素类药物[J]. 分析化学,2012,40(1):101-106. ZHANG Feng, XU Chengbao, LAN Fang, et al. Simultaneous determination of nine androgen drugs in feeds by ultra-performance liquid chromatography-quadrupole-time-of-flight mass spectrometry[J]. Chinese J Anal Chem, 2012, 40(1): 101-106(in Chinese). [19]CHEN F, YING G G, KONG L X, et al. Distribution and accumulation of endocrine-disrupting chemicals and pharmaceuticals in wastewater irrigated soils in Hebei, China[J]. Environmental Pollution, 2011, 159(6): 1 490-1 498. [20]LIU J L, WONG M H. Pharmaceuticals and personal care products (PPCPs): A review on environmental contamination in China[J]. Environment International, 2013, 59(3): 208-224. [21]PENG X Z, OU W H, WANG C W, et al. Occurrence and ecological potential of pharmaceuticals and personal care products in groundwater and reservoirs in the vicinity of municipal landfills in China[J]. Science of the Total Enviroment, 2014, 490: 889-898. [22]XU J, WU L S, CHEN W P, et al. Simultaneous determination of pharmaceuticals, endocrine disrupting compounds and hormone in soils by gas chromatography-mass spectrometry[J]. J Chromatogr A, 2008, 1 202(2): 189-195. [23]KOTNIK K, KOSJEK T, KRAJNC U, et al. Trace analysis of benzophenone-derived compounds in surface waters and sediments using solid-phase extraction and microwave-assisted extraction followed by gas chromatography-mass spectrometry[J]. Anal Bioanal Chem, 2014, 406(13): 3 179-3 190. [24]张长,曾光明,余健,等. 双酚A在湘江沉积物上的吸附特征[J]. 中国环境科学,2006,26(5):550-554. ZHANG Chang, ZENG Guangming, YU Jian, et al. Sorption characteristics of bisphenol A on Xiangjiang river sediments[J]. China Environ Sci, 2006, 26(5): 550-554(in Chinese). [25]ZHANG T, SUN H W, GERECKE A C, et al. Comparison of two extraction methods for the analysis of per- and polyfluorinated chemicals in digested sewage sludge[J]. J Chromatogr A, 2010, 1 217(31): 5 026-5 034. [26]王道玮,赵世民,金伟,等. 加速溶剂萃取-固相萃取净化-气相色谱/质谱法测定沉积物中多氯联苯和多环芳烃[J]. 分析化学,2013,41(6):861-868. WANG Daowei, ZHAO Shimin, JIN Wei, et al. Simultaneous determination of 28 polychlorinated biphenyls and 16 polycyclic aromatic hydrocarbons in sediments using ASE-SPE-GC-QqQ-MS/MS[J]. Chinese J Anal Chem, 2013, 41(6): 861-868(in Chinese). [27]PENG X, YU Y, TANG C, et al. Occurrence of steroid estrogens, endocrine-disrupting phenols, and acid pharmaceutical residues in urban riverine water of the Pearl River Delta, South China[J]. Science of the Total Environment, 2008, 397(1/2/3): 158-166. Determination of Seven New Emerging Pollutants in Sediment Using UPLC-QTOF-MS TAN Jian-hua1,2, TANG Jia-jun1, OUYANG Pei-yu1, WANG Ying-li1, XIE Qi-lai1, YANG Yu-xiang1, CHEN Zhen-xin1, LIU Xin-yu1,3 (1.KeyLaboratoryofSoilEnvironmentandWasteReuseinAgriculture ofGuangdongHigherEducationInstitutes,CollegeofNaturalResourcesandEnvironment ofSouthChinaAgriculturalUniversity,Guangzhou510642,China; 2.GuangzhouQualitySupervisionandTestingInstitute,Guangzhou510110,China; 3.ThePearlRiverWaterEnvironmentMonitoringCenter,Guangzhou510611,China) Abstract:A method of ultra-high performance liquid chromatography coupled with quadrupole time-of-flight high resolution mass spectrometry (UPLC-QTOF-MS) was developed for the determination of seven emerging pollutants in sediment, such as salicylic acid, naproxen, ibuprofen, ketoprofen, diclofenac, clofibric acid and triclosan. Sample was extracted with ultrasonic-assisted treatment using the mixture of formic acid-acetone (1∶99,V/V) as extraction solvent, and then purified with solid-phase extraction using a mixed-mode anion exchange (MAX) cartridge, and finally detected by UPLC-QTOF-MS equipped with an electrospray ionization (ESI) source operated in negative mode. Qualitative screening was achieved according to the obtained experimental results with respect to mass accuracy, isotope distribution and relative abundance of the selected ions, i.e, a quasi-molecular ion and a fragment ion for each analyte, and retention time of the corresponding targeted compound. The positive samples, found to contain some/all of the targeted pollutants, were subject to MS/MS scan using the quasi-molecular ions of the analytes as precursor ions, thus generating fragmental ions for further confirmation. Quantification was performed with peak areas of extracted ions within a mass window of 0.05 Da. Satisfactory linearities (R2>0.99) are obtained for all analytes in the concentration range of 2-500 μg/L. Limits of detection (S/N=3) and limits of quantification (S/N=10) are in the range of 0.5-0.6 μg/kg and 1.5-2.0 μg/kg (dry weight (DW)), respectively. Recoveries are 87.8%-105.1% at three spiking levels with relative standard deviations (n=5) from 3.1% to 12.2%. The developed method was applied to the analysis of the investigated compounds in surface sediment samples collected from the Liuxi River in Guangzhou, and 4 pollutants are found with the highest concentration range of 6.51-12.26 μg/kg (DW). This method is efficient, accurate, and suitable for analysis of the seven investigated emerging pollutants in sediments. Key words:ultra-high performance liquid chromatography quadrupole time-of-flight high resolution mass spectrometry (UPLC-QTOF-MS); sediment; emerging pollutant; solid phase extraction (SPE) 通信作者:解启来(1964—),男(汉族),安徽人,博士生导师,从事环境有机污染物检测技术研究。E-mail: xieql@scau.edu.cn 作者简介:谭建华(1982—),男(汉族),湖南人,高级工程师,从事色谱-质谱检测技术研究。E-mail: tanjianhua0734@aliyun.com 基金项目:广东省科技计划项目(2014A020216035)资助 收稿日期:2015-06-15;修回日期:2015-08-06 中图分类号:O657.63 文献标志码:A 文章编号:1004-2997(2016)01-0052-08
猜你喜欢
海洋通报(2022年2期)2022-06-30 06:07:04
海洋石油(2021年3期)2021-11-05 07:43:12
食品安全导刊(2021年20期)2021-08-30 06:39:48
河北环境工程学院学报(2021年1期)2021-03-19 08:43:00
中国蜂业(2018年4期)2018-05-09 06:25:08
当代化工研究(2016年6期)2016-03-20 16:21:46
当代化工研究(2016年5期)2016-03-20 16:21:35
环境科技(2015年4期)2015-11-08 11:10:44
特产研究(2014年4期)2014-04-10 12:54:22
Sciences in Cold and Arid Regions(2014年6期)2014-03-31 00:28:31
