
世界卫生组织(WHO)的体外诊断试剂产品资格预审项目
2016-02-09 06:28吕宏光
中国医疗器械杂志 2016年4期
吕宏光
艾森生物(杭州)有限公司,杭州市,310030
世界卫生组织(WHO)的体外诊断试剂产品资格预审项目
【作者】吕宏光
艾森生物(杭州)有限公司,杭州市,310030
该文概括性地介绍了世界卫生组织对体外诊断试剂产品的资格预审的大致过程、方法和程序,旨在为中国医疗器械制造商申请世界卫生组织的体外诊断试剂资格预审时提供指引,提高申请速度和效率。
世界卫生组织;体外诊断试剂;资格预审;质量体系
0 引言
世界卫生组织(World Health Organization, WHO)资格预审项目,其主旨是为了确保体外诊断产品、药物、高负担疾病的疫苗和免疫相关设备和器械符合全球质量标准,确保安全性和有效性,优化卫生资源的使用和改善人类的健康状况。
该项目由世界卫生组织基本健康技术部(Essential Health Technology, EHT)下设的诊断和实验室技术组(Diagnostics and Laboratory Team, DLT)整体协调和实施,通过标准化的程序对提交申请的产品进行全面的评价。资格预审过程是一个透明的、科学合理的评估过程,主要过程包括档案审查、一致性测试、性能评价和制造商现场检查。资格预审的信息,与其他采购标准结合,最终由联合国和其他采购代理机构用来对体外诊断试剂(IVD)、药物和/或疫苗做出购买决定。
世界卫生组织资格预审的诊断试剂项目,其目的是获得有质量保证的,适合在资源有限的环境下负担得起的体外诊断产品。该项目提供了世界卫生组织成员国、联合国和其他合作机构的技术信息和对现有的艾滋病毒/艾滋病、疟疾、乙型和丙型肝炎等的测试工具和技术质量的建议。
体外诊断试剂资格预审项目是确保在联合国采购时,公共款项以成本效益最佳的方式进行消费。
截至2014年1月8日,世界卫生组织收到的体外诊断产品资格预审的申请总计201件,产品和方法学分布见表1[1]:
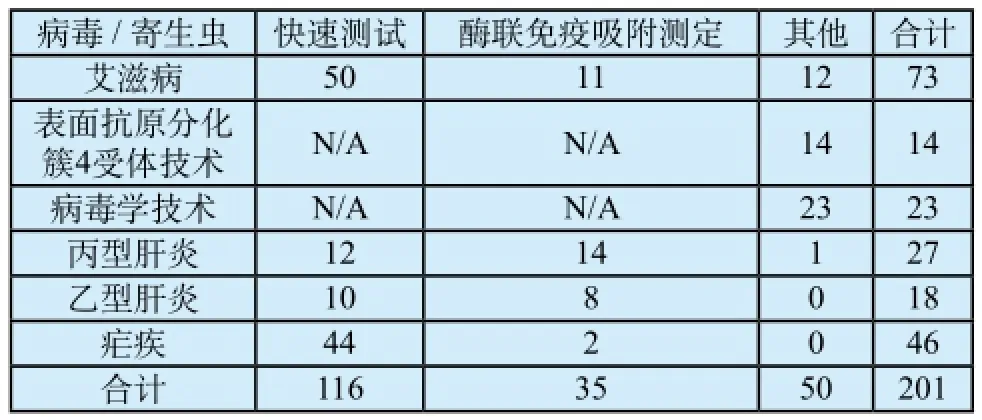
表1 WHO收到的IVD 产品资格预审申请项目分布Tab.1 Distribution of IVD prequalification application received by WHO
1 体外诊断试剂资格预审基本过程
体外诊断试剂资格预审主要包括准备申请表格和产品文档评审、产品实验室评价和制造商现场检查三个部分,共六个主要步骤:提交申请表、制造商签署同意付费书、产品文档评审、制造商现场检查、产品实验室评价、完成资格预审。
1.1 资格预审的申请[2-3]
申请表格的提交是资格预审评估过程的第一步。申请表格中,应提供制造商和体外诊断试剂产品的概要信息。 WHO目前正在接受艾滋病、疟疾、乙型肝炎和丙型肝炎等检测试剂的厂家资格申请。
申请表中除了通常要求提供的制造商和所申请产品的基本信息,如:制造商名称、地址、授权联系人、产品预期使用目的、使用方法、储存条件、有效期等以外,还要求提供制造商和产品的进一步信息,如:产品的法规符合性情况、产品历史、制造商质量体系以及ISO 13485认证情况、关键供应商等信息。
对于CD4 技术(CD4 Technologies )和病毒学技术(Virological Technologies)的体外诊断产品,还提出了更具体的要求。
1.2 制造商签署同意付费书
如果产品符合联合国的优先标准,并准备进行下一步的世界卫生组织资格预审评估过程,该结果会通知到制造商。随后,世界卫生组织将发送一个正式的协议书给制造商,该文件要求制造商签字后回传世界卫生组织,并支付资格预审评估费12 000美元,完成这个过程,制造商将收到提交产品档案的邀请 。
预审评估费用只是部分的由制造商承担,并非全部的费用,因此,即使资格预审项目最终没有通过,这些费用也是不退回制造商的。 这些费用主要用于产品档案审查,产品的实验室评价,制造现场检查,对资格预审评估报告发布等过程。
制造商应特别注意,根据资格预审评估结果,世界卫生组织有权决定,产品是否符合要求并通过资格预审。因此,对资格预审评估费支付并不必然保证所申请产品将通过资格预审。如果制造商不能,或未能在规定的时间内提供所需的信息,或者提供的信息不足以有效地完成预审评估,世界卫生组织也有权在任何阶段终止资格预审评估过程。
同时,世界卫生组织认为如果对于制造商的一个变更的评估是必需的,制造商可能需要支付额外的变更评估费。这将根据具体的案例进行决定。
1.3 产品档案审查
产品档案审查,目的是获得对申请产品的了解,以及理解产品是如何工作的;同时增加了产品制造过程的理解,确定该产品适用于WHO成员国;确定产品是否已经具备实验室评价和制造商现场检查的条件。
制造商应特别注意,除非世界卫生组织明确指示,不应提交产品档案或支付资格预审评估费 ,否则产品档案将未经评审而退回到制造商。
产品档案评估结果包括(但不限于)提交的文件和数据的不充分,WHO会以书面形式与申请人沟通,适当时,要求提交补充数据和信息。
产品档案审查的内容,是庞大而复杂的,主要包括以下信息:
(1) 产品档案形式审查;
(2) 产品基本信息,包括法规符合状况、基本要求符合性检查表、风险分析和控制措施概要等;
(3) 产品设计和制造信息,包括产品形式和组件说明、生物安全、变更历史、制造过程描述、主要原材料和关键供方信息等;
(4) 产品性能标准、相关的验证和确认,包括分析性能研究(如样本类型、分析灵敏度、分析特异性、校准和控制材料的量值溯源性等);
(5) 稳定性研究(产品有效期确定、使用中的稳定性(in-use stability)和运输稳定性等);
(6) 软件的验证和确认;
(7) 临床证据;
(8) 产品标签、标识;
(9) 产品的法规符合性历史;
(10) 产品的上市历史;
(11) 质量管理体系信息等。
通过审查,如果发现申请文件/产品档案不完整,制造商将被要求在指定的时间内提交补充信息。一旦申请文件被接受,世界卫生组织将安排进行产品实验室评价和制造商现场检查。
1.4 现场检查[4]
制造商的生产现场检查,其目的是评估制造商的质量管理体系的充分性、有效性和文件化程序的正确执行。现场检查是基于国际公认的标准,由世界卫生组织的检查员和外聘检查员进行,制造商所在的国的医疗器械主管当局将作为观察员被邀请。通常,现场检查的依据是ISO 13485:2003《医疗器械-质量管理体系-用于法规的要求》[5]标准和ISO/TR 14969 《医疗器械- 质量管理体系- ISO 13485:2003应用指南》[6]。 检查的范围通常限定在制造商确定的生产场所和所申请的产品,如果同时申请几个产品的资格预审,现场检查可以覆盖一个以上的产品。检查将涉及制造商的所有与所申请产品相关的活动和过程。检查小组将审查相关的质量管理体系和产品生产文件,包括标准操作程序和记录。
检查组会事先与制造商沟通,确保现场检查时,制造商必须有至少一个所申请产品的生产活动,此外,在检查时制造商的关键人员必须在场。
检查将被限制在确定的时间内完成。如果检查中,确定有重大的不符合项,那么检查组会与制造商讨论确定,再次检查可能是必要的。
1.5 产品的实验室评估申请
在产品档案审查顺利通过以后,将进行产品的实验室评价,主要针对产品的操作性和性能进行评价,该评价活动由指定的世界卫生组织合作实验室执行。
世界卫生组织认为,对产品质量和性能、以及商业上可用的检测工具和技术的评价是一个全球性的需求,然而,在许多国家法规监管能力是有限的。近年来,越来越多的诊断产品,生产和市场销售行为没有遵循一个有效的许可和监管程序,这是一个非常值得关注的问题。
在实验室的评价前,评价方案将被转发给制造商。方案概述了产品性能和操作特性评估的程序,实验室评价所需的产品数量将取决于所申请的评估产品/被测物。
只有在世界卫生组织的指导下,制造商才可以联系世界卫生组织合作实验室,以便提供足够数量的诊断产品。必要时,提供执行实验室评价所需的设备。
需要特别注意的是,除非得到世界卫生组织合作实验室的正式通知,制造商不应该把诊断产品送往合作实验室,否则,这些产品将被销毁。
合作实验中心应提交一份评估报告草案给世界卫生组织。在评审后,世界卫生组织将报告草案给制造商,使制造商有机会审查实验室评价结果。 但世界卫生组织,对评价结果保持充分的控制权和出版权利。
实验室评估通常包括以下项目:
(1) 测试类型
简单/快速;酶联免疫吸附试验(ELISA);验证性因素分析(如免疫印迹,免疫检测线);CD4 +计数技术;HIV病毒载量监测。
(2) 产品的操作特性
灵敏度;特异性;操作方便性;储存条件。
实验室评价结果以综合报告的形式定期公布,以方便健康卫生政策的制定者、血库管理者、国家艾滋病预防和监测项目管理人员使用。实验结果也被世界卫生组织用来选择和分析大批量的产品采购。
1.6 结果
经过产品档案评审、产品实验室评估和制造商质量体系的现场检查,如果资格预审的所有要求均符合,就可以成为合格的联合国采购招投标制造商。
2 资格预审评估结果
当世界卫生组织对诊断试剂的资格评价结果感到满意并确认了该诊断试剂满足世界卫生组织的所有资格预审要求,该制造商在其确定的生产制造场所生产的通过资格预审的该诊断试剂,将被列入世界卫生组织的资格预审器械清单,并在世界卫生组织的网站上公布;在该清单中,通常包括“资格预审年份、诊断类型、器械名称、器械编号、适用的器械法规、制造商名称、制造场所、包装形式”等内容。
针对通过资格预审的诊断器械、制造商,世界卫生组织会指定一个资格预审编号(Prequalification(PQ) number),通过世界卫生组织资格预审的制造商和诊断器械,就具备参与联合国的采购资格。
诊断试剂制造商将会收到一封世界卫生组织的通知信件,告知其总体的评价结果;当通过资格预审的诊断试剂被列入世界卫生组织的清单中后,制造商应负责将该诊断试剂的生产过程、生产场地和质量控制等方面的变更通知到世界卫生组织。同时,制造商应建立投诉处理程序,并通知世界卫生组织该诊断试剂的任何重大投诉和处理结果。
通过世界卫生组织的资格预审,并不意味着该器械通过了世界卫生组织批准、认证和获得许可;因为对于诊断器械的批准、注册和认可是各个国家和地区医疗器械主管当局的责任,而不是世界卫生组织。
世界卫生组织负责整个的诊断试剂的评价过程和程序,所有的评价报告所有权归世界卫生组织;但世界卫生组织承诺确保诊断试剂制造商的商业敏感信息的保密性;制造商应该明白世界卫生组织也会公布整个评价过程中的信息,如:器械名称、参与资格预审的制造商名称、评审的进展状态、产品档案评审的总结性报告、实验室评价结果和现场审核发现项的总结性报告等。
同时,世界卫生组织保留与联合国和相关国家医疗器械主管当局分享完整的评价和检查报告的权利。
3 资格预审器械的上市后监督
世界卫生组织建立和完善上市后的监督系统,以确保通过预审的器械持续符合世界卫生组织的资格预审要求;上市后的监督系统包括主动收集上市后的器械的质量、安全和性能信息;也包括该器械的不良事件报告、相关主管当局的警戒系统等;
通过资格预审器械的制造商,应该采取如下上市后的监督活动:
(1) 通知世界卫生组织,该器械的所有不良事件以及与该事件有关的详细信息;
(2) 通知世界卫生组织,所有的安全纠正措施(Field safety corrective action)信息,如产品召回、变更等;
(3) 适当时,免费提供给世界卫生组织或其指定实验室足够数量的器械,用于上市后监督测试之用。
任何与资格预审器械有关的事件/投诉,世界卫生组织将根据所建立的标准操作程序(SOP)进行调查;并根据事件、投诉的类型和程度,通知制造商和相关国家/地区的医疗器械主管当局。
4 资格预审的进度跟踪
世界卫生组织在其网站上定期公布每一个申请产品的资格预审进展情况,以便于申请者了解整个项目的进展情况。
在世界卫生组织的网站上,参与资格预审的制造商和相关方,可以根据以下信息,查询项目的进展,指示符号代表的过程阶段如表2所示。

表2 指示符号代表的过程阶段Tab.2 Symbolic representation of the process stage
5 资格预审中的其他注意事项
5.1 资格预审产品的变更通知
通过资格预审的诊断试剂和制造商,如果产品、生产场所或生产过程发生变化,则通过资格预审的诊断试剂可能就不符合世界卫生组织的资格预审要求,制造商应将变更及时通知世界卫生组织;世界卫生组织将根据建立的标准操作程序(SOP)确认这些变更的影响程度,并确定后续的处置措施。
(1) 通过对制造商提供的变更信息的评审,认为变更是可以接受的;
(2) 对资格预审的诊断器械进行追加评审;
(3) 重大变更,导致产品功能、性能实质性变化,世界卫生组织认为应按新产品进行资格预审。
世界卫生组织将根据评审的结果,建议制造商如何进行这些变更的处理
5.2 关于“贴牌(re-branding)”器械的安排
世界卫生组织知道,某些公司会购买其它制造商的成品器械,并贴加上自己的品牌。世界卫生组织认为“贴牌(re-branding)”产品应是与原始制造商在相同的制造条件、在同一生产场所生产的同质的产品。换句话说,一个“贴牌(re-branding)”产品是在各方面与原始制造商相同的器械,除了产品标有“不同品牌”的名称和标识符。
世界卫生组织鼓励原始制造商和贴牌制造商联合进行资格申请。但其条件是申请者明确同意世界卫生组织可以公开披露该器械为“贴牌产品”。
6 资格预审状态的有效性
世界卫生组织将在确定的周期内安排对列入资格预审名单的产品和制造场所进行再评价,如果再评价结果表明,资格预审器械和制造商不再符合世界卫生组织的资格预审要求,则器械和制造场所将从清单中移除。同样地,如果制造商不能积极参与和配合世界卫生组织的再评价程序,也将从清单中被移除。
7 总结
综上所述,可以明确的是,世界卫生组织的诊断试剂资格预审程序不同于产品质量认证和质量体系认证,也有别于医疗器械主管当局的上市前批准程序,即使在操作上与某些国家和地区的上市前批准程序有类似之处。
世界卫生组织的诊断试剂资格预审程序有其自身的独立性和程序的独特性;评价过程严格、评价周期长;同时,参与该申请过程的制造商也要充分评价和认识到该程序对企业运行和产品的影响程度,因为一些评价信息是会在世界卫生组织的网站上公布的。
世界卫生组织的诊断器械资格预审程序,对医疗器械(诊断试剂)的安全性和有效性无疑是多了一层的保障。制造商应该明了,该资格预审过程漫长,通常需要3~4年的时间。制造商要充分评估企业的自身实力,再决定是否进入资格预审程序。
[1] PQDx_007 v4 Overview of prequalification of in vitro diagnostics[R/OL]. [2011-03-22]. http://www.who.int/diagnostics_ laboratory/evaluations/140530_pqdx_overview_doc_007.
[2] PQDx_015 v3 Application form[R/OL]. [2010-02-02]. http://www. who.int/diagnostics_laboratory/evaluations/Application/en/.
[3] PQDx_017 v4 Instruction for the completion of the prequalificaiton of in vitro diagnostic pre-submission form[R/OL]. [ 2014-03-30]. http://www.who.int/diagnostics_laboratory/evaluations/140530_ pqdx_017_psf_instructions_v4_final.pdf?ua=1.
[4] PQDx_014 v3 Information for Manufacturers on Inspections Prequalification of Diagnostics[R/OL]. [2010-03-30]. http://www. who.int/diagnostics_laboratory/evaluations/140717_pqdx_014_ info_manufacturers_pq_inspections_final.pdf ua=1.
[5] ISO 13485: 2003 医疗器械 质量管理体系 用于法规的要求[S].
[6] ISO/TR 14969 医疗器械- 质量管理体系- ISO13485: 2003 应用指南[S].
WHO in Vitro Diagnostic Reagent Products Prequalifcation Program
【Writer】LV Hongguang
ACEA Biosciences (Hangzhou) Co. Ltd., Hangzhou, 310030
This paper briefly introduces the WHO for prequalification of in vitro diagnostic reagents process, methods and procedures, provides guidelines for prequalification to apply for in vitro diagnostic reagents WHO for Chinese manufacturers of medical devices, applications for improving the speed and efficiency.
WHO, in vitro diagnostic reagents, prequalification, quality system
F203
A
10.3969/j.issn.1671-7104.2016.04.011
1671-7104(2016)03-0275-04
2016-01-29
吕宏光,E-mail:hongguang.lv@aceabio.com.cn
猜你喜欢
英语文摘(2020年2期)2020-08-13
户外探险(2020年3期)2020-04-01
江西社会科学(2019年10期)2019-11-11
铁道警察学院学报(2019年4期)2019-10-31
少年文艺·开心阅读作文(2017年4期)2017-04-07
中国工程咨询(2017年10期)2017-01-31
小说月刊(2015年9期)2015-04-23
——侦查程序中不容回避的理论和现实问题
中国刑警学院学报(2015年4期)2015-01-29
小说月刊(2014年11期)2014-11-18
知识窗(2009年10期)2009-12-01
