
动物性食品中泰妙菌素残留标志物检测UPLC-MS/MS法研究
2016-02-07 08:05:17叶妮尹晖王亦琳孙雷王鹤佳
中国兽药杂志 2016年11期
叶妮,尹晖,王亦琳,孙雷,王鹤佳
(中国兽医药品监察所,北京 100081)
动物性食品中泰妙菌素残留标志物检测UPLC-MS/MS法研究
叶妮,尹晖,王亦琳,孙雷*,王鹤佳
(中国兽医药品监察所,北京 100081)
建立了猪和鸡的肌肉、肝脏以及鸡皮+脂组织中泰妙菌素残留标志物(8-α-Hydroxymutilin)残留检测的UPLC-MS/MS方法。样品经酸化丙酮提取,氢氧化钠水解,正己烷除脂,二氯甲烷反萃取后,用C18色谱柱分离,以0.1%甲酸乙腈溶液和0.1%甲酸水溶液为流动相进行梯度洗脱,基质添加标准溶液外标法定量。结果表明:8-α-Hydroxymutilin在20~500 μg/kg猪肌肉、鸡肌肉和鸡皮+脂基质添加标准溶液浓度范围、100~2000 μg/kg猪肝基质添加标准溶液浓度范围、200~4000 μg/kg鸡肝基质添加标准溶液浓度范围内呈现良好线性关系,相关系数R2均大于0.990;方法定量限:猪肌肉、鸡肌肉、鸡皮+脂为25 μg/kg,猪肝脏为100 μg/kg,鸡肝脏为250 μg/kg。猪肌肉、鸡肌肉、鸡皮+脂在25~200 μg/kg添加浓度范围内、鸡肝脏在250~2000 μg/kg添加浓度范围内、猪肝脏在100~1000 μg/kg添加浓度范围内,8-α-Hydroxymutilin的回收率范围均为60%~120%,批内、批间相对标准偏差均小于20%,满足国内外兽药残留相关法规规定。
动物性食品;泰妙菌素残留标志物;残留;超高效液相色谱-串联质谱
泰妙菌素是一种截短侧耳素类动物专用抗生素,主要用于防治畜禽呼吸系统疾病和促生长,是广泛应用的兽医专用抗生素之一。该药在动物体内吸收迅速,体内分布广泛,抗菌活性强,对支原体、细菌及螺旋体,如猪肺炎支原体、猪滑液支原体、鸡败血支原体、链球菌、金黄色葡萄球菌、放线杆菌及猪痢疾短螺旋体等都具有良好的抗菌活性[1]。低剂量的泰妙菌素可以促进动物生长,提高饲料利用率,因此作为兽药和饲料添加剂被广泛应用[2]。但是,泰妙菌素残留可能对人类健康造成严重的潜在危害。泰妙菌素在动物体内代谢成8-α-Hydroxymutilin以及各种酯代谢物[3]。据报道,动物组织内8-α-Hydroxymutilin残留量与泰妙菌素总量有关联[4],因此,通过测定动物体内8-α-Hydroxymutilin残留量即可推导计算泰妙菌素的总量。目前我国与欧盟等国均已批准了泰妙菌素在猪、鸡等动物组织中的最大残留限量(MRL),在鸡蛋中的残留标志物为原型,在猪肝脏、肌肉和鸡肝脏、肌肉及皮+脂中的残留标志物为可水解为8-α-Hydroxymutilin代谢产物之和。8-α-Hydroxymutilin在猪肝脏中的MRL为500 μg/kg,在猪肉中的MRL为100 μg/kg,在鸡肝脏中的MRL为1000 μg/kg,在鸡肉和鸡的皮+脂中的MRL为100 μg/kg[4]。目前,我国尚未发布以8-α-Hydroxymutilin为标志物的泰妙菌素残留检测方法标准,已发表的相关文献,均以泰妙菌素原型为残留标志物,与兽药最高残留限量标准规定不符。因此,本试验进行了猪的肌肉、肝脏以及鸡的肌肉、肝脏、皮+脂组织中8-α-Hydroxymutilin残留检测的UPLC-MS/MS方法研究。
1 材料与方法
1.1 仪器 Acqutiy UPLC-Premier XETM质谱联用仪,Waters公司;电子天平,Mettler Toledo 公司;高速冷冻离心机,Heraeus公司;氮吹仪,Jnc公司;涡旋混合器、水平振荡器,IKA 公司;旋转蒸发仪,Buchi公司。
1.2 药品和试剂 8α-羟基-泰妙菌素,纯度99.0%,SANDOZ公司生产,礼来公司提供;甲醇、乙腈、甲酸为色谱纯,Fisher公司;丙酮、正己烷、二氯甲烷、盐酸、氢氧化钠均为分析纯;所用水为超纯水。
1.3 标准溶液配制 精密称取8-α-Hydroxymutilin标准品约10 mg,于10 mL容量瓶中,用丙酮溶解并定容至刻度,配制成1 mg/mL的8-α-Hydroxymutilin标准储备液。准确吸取0.1 mL标准储备液至另一10 mL容量瓶中,用甲醇+0.1%甲酸水溶液(20+80,V/V)溶解并稀释至刻度,配制成10 μg/mL标准工作液。
1.4 样品前处理
1.4.1 提取与水解 准确称取(2±0.02)g试料于50 mL离心管内,加入丙酮-0.5 mol/L盐酸溶液(60∶1,V/V)20 mL,涡旋混匀,中速水平振荡5 min,8000 r/min离心8 min,取上清液于50 mL鸡心瓶中。依次加水5 mL、0.5 moL/L盐酸1 mL,混匀,45 ℃下旋转蒸至无沸腾现象,依次加入水1.5 mL、7 moL/L氢氧化钠溶液0.5 mL,混匀,45 ℃水浴20 min后,取出放置至室温备用。
1.4.2 除脂与萃取 备用液转移至50 mL离心管内,加正己烷10 mL,涡旋混匀,于4 ℃下5000 r/min离心5 min,弃上层液,依次加入水5 mL、浓盐酸0.5 mL和二氯甲烷10 mL,涡旋混匀,于4 ℃下5000 r/min离心5 min,取下层液于10 mL离心管中,45 ℃氮气吹至近干。残余物中加入甲醇+0.1%甲酸水溶液(20+80,V/V)1.0 mL,充分涡旋溶解后转移至1.5 mL离心管中,12000 r/min离心5 min,取适量上清液过0.2 μm滤膜,供超高效液相色谱-串联质谱仪测定。
1.5 仪器条件
1.5.1 色谱条件 色谱柱为BEH C18(50 mm×2.1 mm,1.7 μm);流动相A相为0.1%甲酸乙腈溶液,B相为0.1%甲酸水溶液,梯度洗脱:0.0~5.0 min,10%A线性变化至90%A;5.0~6.5 min,90%A线性变化至10%A;流速为0.3 mL/min;柱温为30 ℃;进样量为10 μL。
1.5.2 质谱条件 电喷雾离子源(ESI+);毛细管电压为3.2 kV;萃取电压为3.0 V;透镜电压为0.5 V;源温为100 ℃;脱溶剂温度为350 ℃;脱溶剂气速为450 L/h;锥孔反吹气速为50 L/h。
1.5.3 定性与定量 该方法定性需满足四个条件:① 空白实验不出现与阳性对照相同的离子峰;② 特征离子色谱峰信噪比(S/N)都在3∶1以上;③ 试样色谱峰保留时间应与校正溶液的一致,容许偏差为±5%;④ 检测到的离子丰度比应与校正溶液的一致,容许偏差达到欧盟2002/657/EC中的规定。定量时以响应强度最强的离子对作为定量离子,通过基质添加标准溶液外标法进行定量。
1.6 定量方法 于空白基质中添加适宜浓度药物进行提取、水解及反萃取后的溶液作为对照溶液进行测定,作为定量依据。1.7 线性测定方法 精密量取8-α-Hydroxymutilin标准工作液适量,制成较高浓度的空白添加试料,和空白试料一起经提取、水解、萃取和吹干后,用甲醇+0.1%甲酸水溶液(20+80,V/V)1.0 mL溶解并过0.2 μm微孔滤膜,制成基质添加试样溶液和空白试样溶液,用空白试样溶液将基质添加试样溶液稀释成含待测药物浓度,猪肝为100、200、500、1000和2000 ng/mL,鸡肝为200、500、1000、2000和4000 ng/mL,猪和鸡肌肉以及鸡皮+脂为20、50、100、200和500 ng/mL系列浓度的基质添加标准溶液,依次上机测定,以特征离子质量色谱峰面积为纵坐标,基质添加标准溶液浓度为横坐标,绘制标准曲线。
2 结果与分析
2.1 监测离子 8-α-Hydroxymutilin属于欧盟指令96/23/EC附录Ⅰ[5]所列的限用物质,根据欧盟2002/657/EC要求,要确证该类物质至少需要3个识别点(IP),因此,我们分别选择了母离子以及对应的两个响应较强的子离子作为定性定量依据,即一母离子1个IP点,对应两子粒子分别1.5个IP点,共4个IP点,完全满足检测方法要求。根据8-α-Hydroxymutilin一级质谱扫描和二级质谱扫描,确定了监测离子(表1)。

表1 8-α-Hydroxymutilin定性、定量离子对及对应锥孔电压、碰撞能量参考值
2.2 定量结果 该方法为针对基质复杂的动物性组织,其前处理步骤较多,且增加了特殊的碱水解和液液反萃取步骤,据相关试验数据表明,水解及液液反萃取步骤会造成约25%的药物损失,并且提取以及浓缩步骤也会使待测物的绝对回收损失,因此,采用了空白基质中添加药物进行提取、水解及反萃取后的溶液作为对照溶液进行定量,即基质添加标准溶液的方式进行定量,可抵消前处理过程中不可避免的损失以及基质效应的干扰,获得了较好的回收率结果。
2.3 线性结果 通过各组织的标准曲线、回归方程及相关系数R2(表2)可以得出:8-α-Hydroxymutilin在有关浓度范围内(猪和鸡肌肉以及鸡皮+脂20~500 ng/mL、猪肝100~2000 ng/mL、鸡肝200~4000 ng/mL)呈现良好的线性关系,R2均大于0.990。
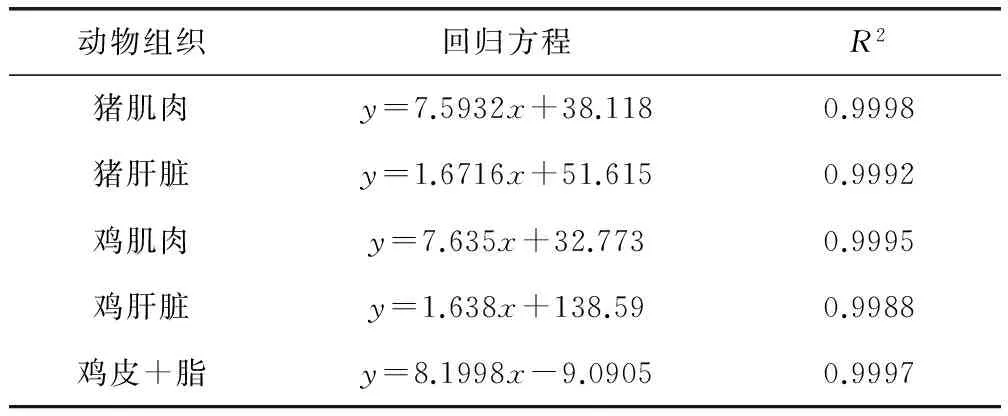
表2 各组织8-α-Hydroxymutilin基质添加标准曲线
2.4 灵敏度和精确度 采用空白组织中添加目标化合物的方法,依据特征离子质量色谱峰信噪比S/N>10为方法定量限,得出8-α-Hydroxymutilin定量限在猪肌肉、鸡肌肉、鸡皮+脂为25 μg/kg;猪肝脏为100 μg/kg;鸡肝脏为250 μg/kg。其中,1000 ng/g空白鸡肝添加试样溶液得到的特征离子质量色谱图见图1。

图1 1000 μg/kg空白鸡肝添加试样溶液特征离子质量色谱图
采用标准添加法,在空白猪、鸡肌肉、肝脏和鸡皮+脂中添加4个不同浓度(定量限、1/2MRL、MRL和2MRL四个浓度)的8-α-Hydroxymutilin进行回收率试验,各浓度进行5个样品平行试验,重复3次,计算批间相对标准偏差。
试验结果表明,本方法对猪肌肉、鸡肌肉、鸡皮+脂在25~200 μg/kg添加浓度范围内、鸡肝脏在250~2000 μg/kg添加浓度范围内、猪肝脏在100~1000 μg/kg添加浓度范围内,8α-羟基-泰妙菌素的回收率范围均为60%~120%。批内、批间相对标准偏差均小于20%。其中,鸡肝组织中8-α-Hydroxymutilin添加回收率实验结果见表3。
3 讨论与结论
本方法与文献方法主要区别在于对样品的前处理过程。由于8-α-Hydroxymutilin为碱性化合物,因此采用酸化丙酮溶液(丙酮∶0.5 moL/mL盐酸)提取组织内的8-α-Hydroxymutilin。由于丙酮的沸点为45 ℃,因此,于此温度下进行旋转蒸发,将提取液中的丙酮和少部分水蒸发,得到浓缩液。猪的肝脏和肌肉以及鸡的肝脏、肌肉和皮肤+脂的残留标志物是可水解为8-α-Hydroxymutilin的代谢物之和。要将8-α-Hydroxymutilin的各种酯代谢物转化成8-α-Hydroxymutilin,需要进行水解。本方法直接参考国外礼来公司提供的碱水解条件,即加入7 mol/L的氢氧化钠溶液,45 ℃水浴中放置20 min的条件,将各种8-α-Hydroxymutilin酯水解成8-α-Hydroxymutilin。
由于动物组织内均存在油脂,影响仪器响应值和信噪比,因此,选用常规去脂试剂正己烷,通过涡旋、4 ℃离心去除油脂。8-α-Hydroxymutilin可很好溶于二氯甲烷,且二氯甲烷具有良好挥发性,因此,选用二氯甲烷作为反萃取溶剂,在酸性条件下从去脂后的溶液中反萃取8-α-Hydroxymutilin,
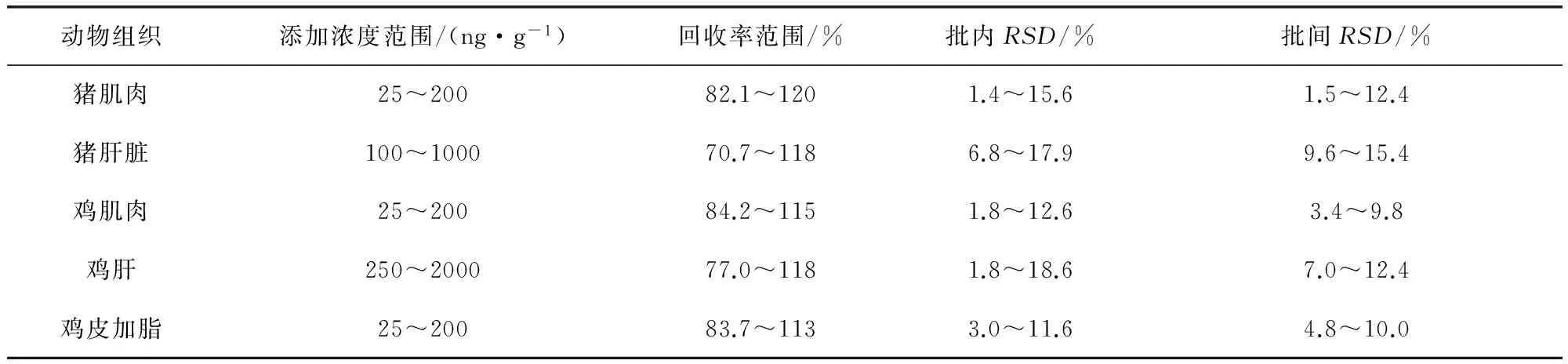
表3 各组织中8-α-Hydroxymutilin添加回收率实验结果(n=5)
于45 ℃氮气吹至近干。由于复溶液中混有沉淀物和悬浮物,使得复溶液呈乳白色,需通过高速离心使其澄清,因此,根据复溶体积(1 mL),选择将复溶液转移至1.5 mL塑料离心管中,以12000 r/min的速度高速离心,吸取澄清部分过0.2 μm滤膜,上机检测。
已有文献部分方法采用HLB萃取柱对样品进行净化,经实验表明,活化萃取柱所用磷酸盐缓冲液的酸性环境可能会造成待测药物的破坏和损失。因此,本实验未采用同类方法。
本方法通过对样品前处理条件的充分研究,建立了适用于猪、鸡的肌肉、肝脏和鸡皮+脂等5种动物性食品中8-α-Hydroxymutilin残留检测的UPLC-MS/MS方法。方法以8-α-Hydroxymutilin为残留标志物,因待测药物为碱性物质,故用萃取、旋蒸和反萃取的方法对样品进行净化,不同于已有
文献方法的以泰妙菌素原型为残留标志物,用有机溶剂提取并经萃取柱柱净化,避免了萃取柱的酸性条件对碱性物质的破坏及损耗,具有良好的可操作性和重现性,方法灵敏度和精确度均能满足兽药残留分析方面的要求,为能够完整的进行动物性食品中泰妙菌素的残留检测提供了重要的技术支持。
[1] 黄贺贤,曾振灵,黄显会. 截短侧耳素类抗生素-泰妙菌素的研究进展[J]. 中国兽药杂志, 2010, 44(6): 42-45.
[2] 马境,王金辉,吴悦泰. 泰妙菌素在兽医临床上的应用[J]. 养殖技术顾问,2007(7):92-92.
[3] 耿士伟,朱志谦. 高效液相-串联质谱法测定鸡肉中泰妙菌素残留的研究[J]. 中国家禽, 2012, 34(11): 28-31.
[4] 农业部公告第235号 动物性食品中兽药最高残留限量[S].
[5] 欧盟指令96/23/EC附录Ⅰ[Z].
(编辑:李文平)
The Research of 8-α-Hydroxymutilin Residues in Animal Derived Food by UPLC-MS/MS
YE Ni, YIN Hui, WANG Yi-lin, SUN Lei*, WANG He-jia
(ChinaInstituteofVeterinaryDrugControl,Beijing100081,China)
A UPLC-MS/MS method was established for the determination of 8-α-Hydroxymutilin residue in animal derived food. After extracted by acid acetone and concentrated, the tissues were hydrolyzed by sodium hydrate solution, removed oil byn-hexane. The separation of 8-α-Hydroxymutilin was performed on Waters Acquity UPLC system with the column of BEH C18 and the gradient elution solvent of acetonitrile (0.1% formic acid) and water (0.1% formic acid) at a flow rate of 0.3 mL/min. The method was quantified by blank tissue spiked standard curves. The calibration curves were good linear between the peak areas and the concentrations of 20~500 μg/kg for pig muscle, chicken muscle and skin and fat of chicken,100~2000 μg/kg for pig liver, 200~4000 μg/kg for chicken liver, the correlation coefficientR2>0.990. The limits of detection of 8-α-Hydroxymutilin in pig muscle, chicken muscle and skin and fat of chicken were 25 μg/kg, in pig liver was 100 μg/kg and in chicken liver was 250 μg/kg. The average recoveries from spiked animal tissues at the concentrations of 25~200 μg/kg for pig muscle, chicken muscle and skin and fat of chicken, 250~2000 μg/kg for chicken liver, 100~1000 μg/kg for pig liver, ranged 60%~120%. The intra- and inter-batch variation coefficients were both less than 20%.
animal derived food;8-α-Hydroxymutilin;residue;UPLC-MS/MS
叶妮,助理研究员,从事兽药残留相关研究。
2016-08-08
A
1002-1280 (2016) 11-0049-05
S859.84
猜你喜欢
广东医科大学学报(2020年4期)2020-08-24 07:11:10
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:06
中成药(2018年12期)2018-12-29 12:25:44
中成药(2017年6期)2017-06-13 07:30:35
广东饲料(2016年6期)2016-12-01 03:43:24
中华老年多器官疾病杂志(2016年9期)2016-04-28 08:52:44
医学研究杂志(2015年7期)2015-06-22 11:01:10
医学研究杂志(2015年4期)2015-06-10 06:42:43
现代检验医学杂志(2015年6期)2015-02-06 01:44:22
现代检验医学杂志(2015年5期)2015-02-06 01:42:23
