
大黄末中没食子酸的鉴别及含量测定方法研究
2016-02-07 07:04蔡文金包爱情陆春波
中国兽药杂志 2016年1期
蔡文金, 包爱情, 陆春波
(浙江省兽药饲料监察所,杭州 310012)
大黄末中没食子酸的鉴别及含量测定方法研究
蔡文金, 包爱情, 陆春波
(浙江省兽药饲料监察所,杭州 310012)
为了完善大黄末的质量控制方法,对大黄末中没食子酸的薄层鉴别和含量测定方法分别进行了研究。采用薄层色谱法鉴别没食子酸以及高效液相色谱法测定其含量。选用Agilent Eclipse XDB-C18 (250 mm×4.6 mm, 5 μm)为色谱柱,甲醇-0.05%磷酸溶液(5∶95)为流动相,流速1.0 mL/min,检测波长为271 nm。试验结果没食子酸在1.064~106.4 μg/mL浓度范围内,线性关系良好(r= 1.0000);平均回收率(n=6)为97.84%(RSD=0.61%)。薄层色谱斑点清晰,分离度好。本方法准确灵敏,可用于大黄末中没食子酸的质量控制。
大黄末;没食子酸;薄层色谱法;高效液相色谱法
大黄末收载于《中国兽药典》二O一O年版二部,为单味大黄制成的中兽药散剂[1]。具有健胃消食,泻热通肠,凉血解毒,破积行瘀功效,用于食欲不振,实热便秘,结症,疮黄疔毒,目赤肿痛,烧伤烫伤,跌打损伤,还可以用于治疗鱼肠炎,烂鳃,腐皮[2]。大黄为常用中药, 主要含有蒽醌类、二苯乙烯苷类、苯丁酮类, 鞣质及相关的多酚类成分。现代药理表明蒽醌类为大黄泻下作用的主要有效成分,没食子酸等鞣质为大黄收敛止血作用的主要有效成分[3]。大黄末现行质量标准中仅有蒽醌类的薄层色谱鉴别和含量测定,无鞣质的鉴别和含量测定。查阅文献,未发现有大黄中没食子酸薄层色谱鉴别的相关报道,本试验参照石榴皮药材中的没食子酸鉴别[1]和含量测定方法[4-5],对大黄末中没食子酸的薄层鉴别和含量测定方法分别进行了研究。
1 材料
1.1 仪器 Waters 2695 高效液相色谱仪,配Waters 2996二极管阵列检测器,美国Waters公司;AUTOMATIC TLC SAMPLER 4全自动点样仪,ADC2自动展开仪和TLC VISUALIZER 薄层色谱成像系统,瑞士KAMAG公司; XS-205电子天平(感量0.01 mg),瑞士METTLER TOLEDO公司;KQ-500E型超声波清洗器,昆山市超声仪器有限公司。
1.2 试剂与材料 甲醇为色谱纯,Merck 公司,其余试剂均为分析纯;实验用水为milli-Q 超纯水。硅胶G薄层板,100 mm×200 mm,厚度0.20~0.25 mm,青岛海洋化工厂生产。没食子酸对照品(中国食品药品检定检验院,批号:110831-201204,含量:89.9%)。大黄末样品(批号分别为20141101,20141102,20141103),浙江歌德动物药业生产;大黄末样品(批号分别为20150301,20150302,20150303,20141203),浙江东贸药业生产。
2 方法与结果
2.1 没食子酸的薄层色谱鉴别 取供试品10 g,加无水乙醇30 mL,加热回流1 h,滤过,取滤液蒸干,加水20 mL使溶解,滤过,滤液用石油醚(60~90 ℃)提取2次,每次20 mL,弃去石油醚液,水液再用乙酸乙酯振摇提取2次,每次20 mL,合并乙酸乙酯液,蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取没食子酸对照品10 mg,置10 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,作为对照品溶液。照薄层色谱法,吸取上述两种溶液各5 μL,分别点于同一硅胶G薄层板上,以环己烷(用水饱和)-乙酸乙酯-甲酸(6:5:1)为展开剂,展开,取出,晾干,喷以1%三氯化铁乙醇溶液。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。斑点清晰,分离度好。结果见图1。
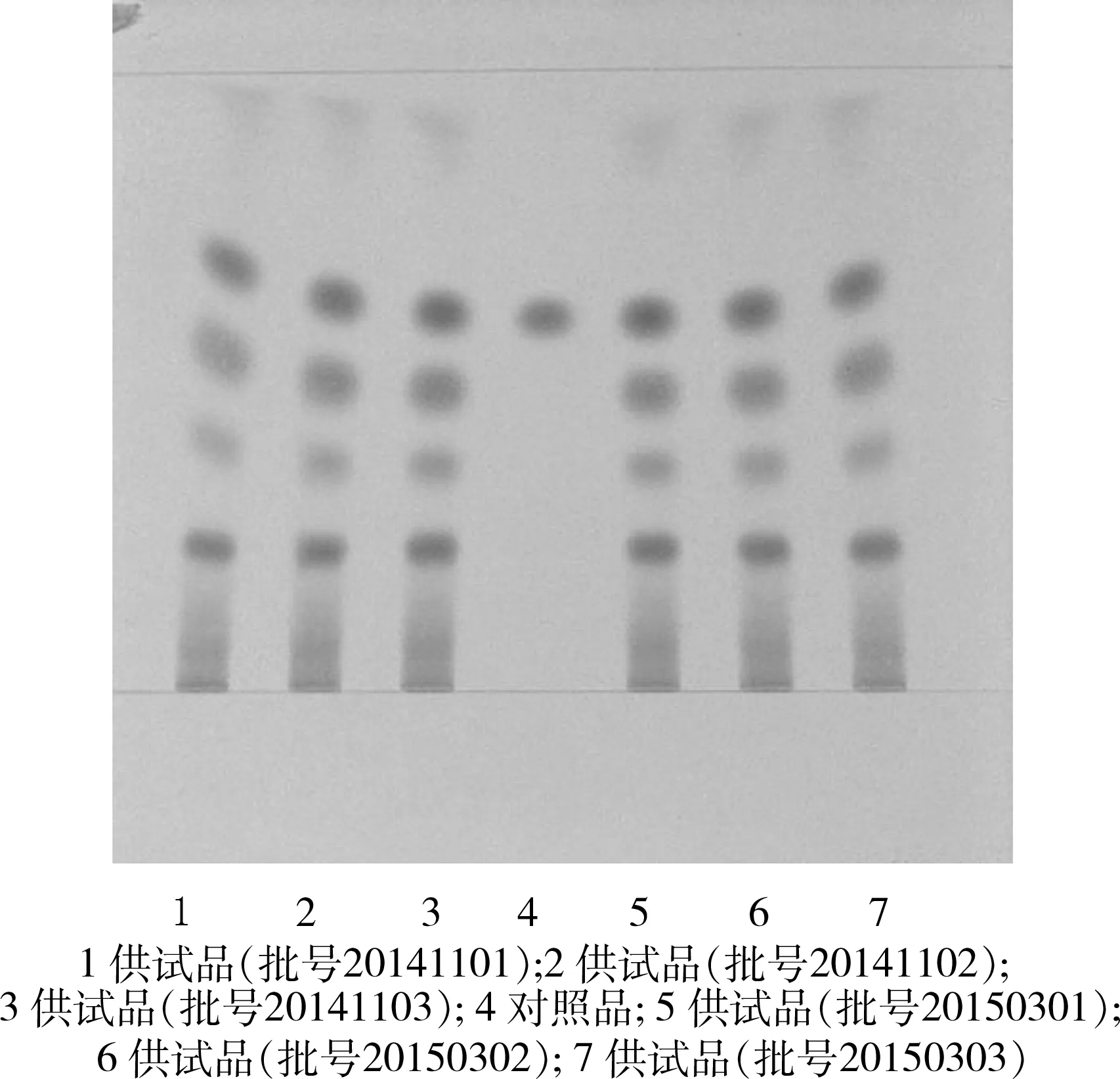
图1 没食子酸薄层鉴别图谱
2.2 没食子酸的含量测定
2.2.1 色谱条件 色谱柱:Agilent Eclipse XDB-C18(250 mm×4.6 mm,5 μm);流动相:甲醇-0.05%磷酸溶液(5∶95);流速1.0 mL/min;采集波长200~400 nm,分辨率为1.2 nm;进样量:20 μL;柱温30 ℃。
2.2.2 对照品溶液的制备 精密称取没食子酸对照品23.68 mg,置100 mL容量瓶中,加水溶解并稀释至刻度,摇匀,为对照品储备液。精密量取上述溶液5 mL,置100 mL量瓶中,用水稀释至刻度,摇匀,即得21.29 μg/mL的对照品溶液。
2.2.3 供试品溶液的制备 取大黄末约0.1 g,精密称定,置25 mL量瓶中,加水20 mL,超声提取30 min,静置30 min,再加水至刻度,摇匀,即得。
2.2.4 系统适应性试验 分别取对照品溶液、供试品溶液注入液相色谱仪,同时记录光谱图和271 nm处的色谱图,分别见图2、图3。在该色谱条件下,理论板数按没食子酸峰计为10403,没食子酸可以得到很好的分离。
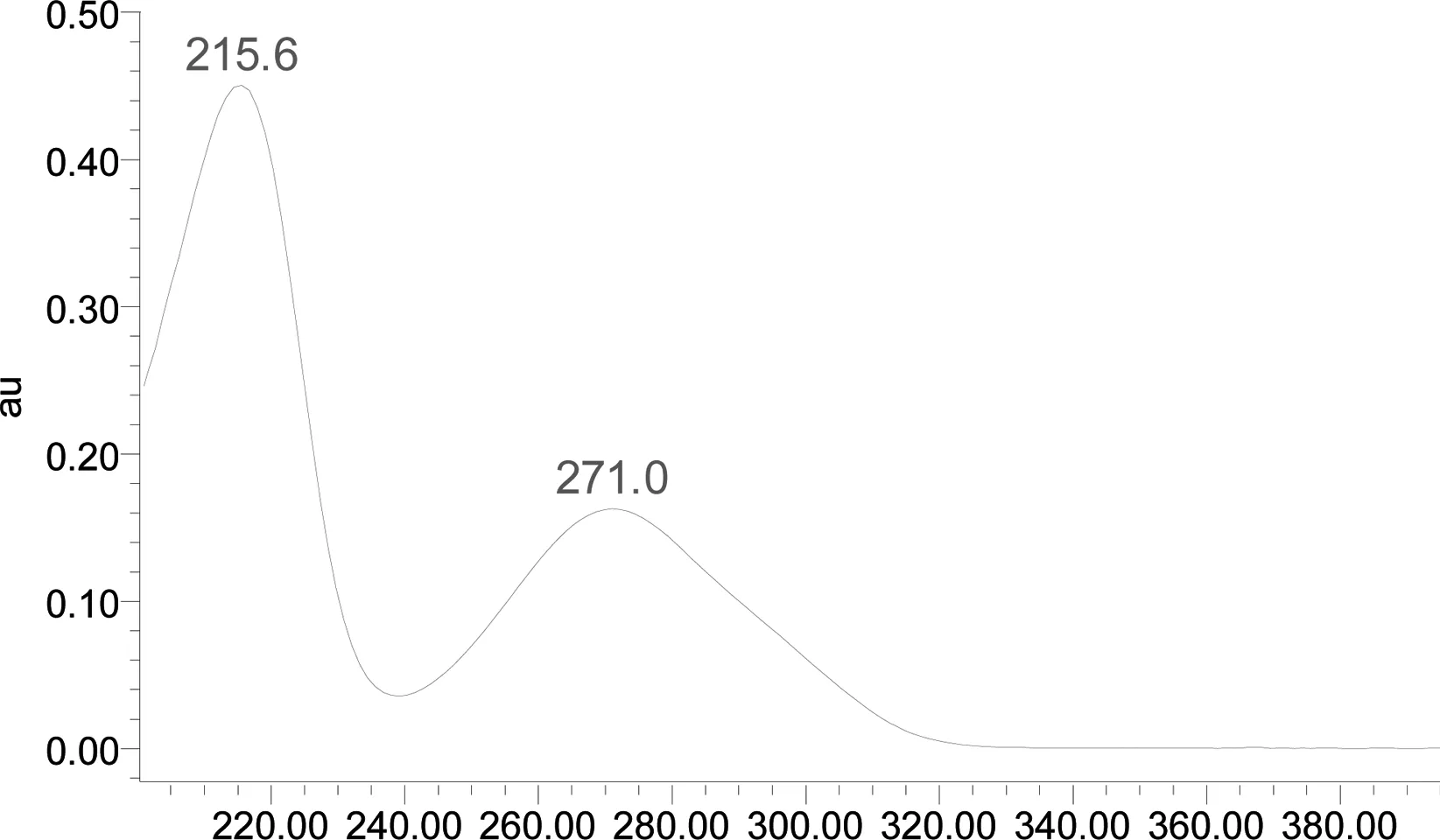
图2 没食子酸光谱图
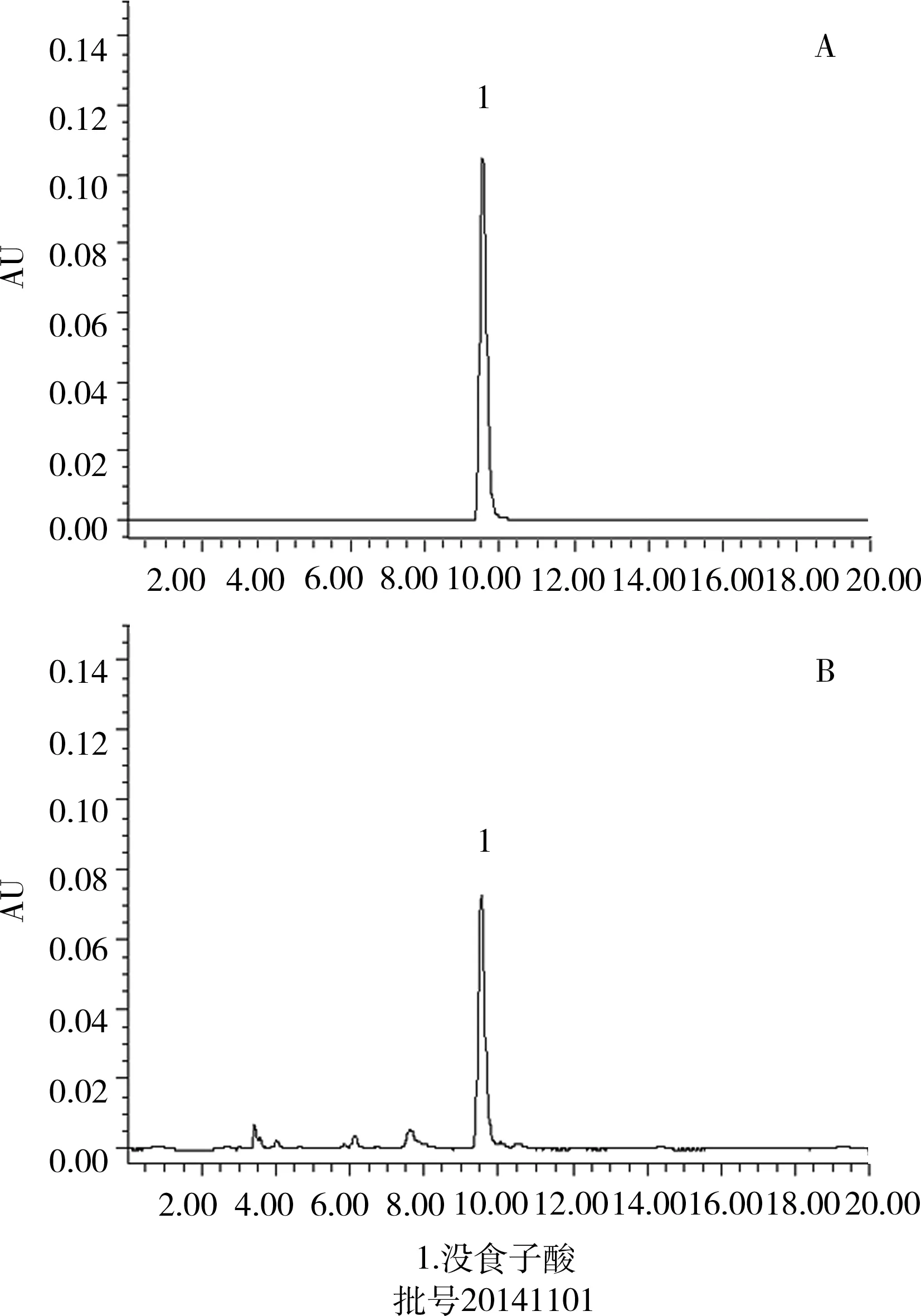
图3 对照品溶液(A)和供试品溶液(B)色谱图
2.2.5 线性关系考察 分别精密量取对照品储备液适量,用水稀释成浓度约为1、2、5、10、20、50、100 μg/mL的系列溶液,精密量取20 μL,注入液相色谱仪,记录色谱图。以浓度(X)为横坐标,峰面积(Y)为纵坐标,绘制标准曲线,所得线性方程为:
Y=64897X-11277(r=1.0000)。结果表明没食子酸对照品溶液在1.064~106.4 μg/mL浓度范围内,线性关系良好。2.2.6 精密度试验 精密量取浓度为53.22 μg/mL的对照品溶液20 μL注入液相色谱仪,按2.2.1色谱条件连续进样6次,记录色谱图。结果没食子酸峰面积的相对标准偏差(RSD)为0.4 %,表明精密度良好。
2.2.7 稳定性试验 取同一批供试品溶液(批号20141103),分别在样品处理后的0、2、4、6、8、12 h进样,测定峰面积相对标准偏差(RSD)为0.8%,表明溶液在12 h内稳定。
2.2.8 重复性试验 取大黄末样品(批号20141103)5份,按“2.2.3”项下的方法制备供试品溶液,分别进样测定含量。结果样品中没食子酸含量的相对标准偏差(RSD)为0.7%,说明方法重复性良好。
2.2.9 加样回收率试验 取“2.2.8”项下已测含量的大黄末样品(批号20141103)0.05 g,共 6份,精密称定,置25 mL量瓶中,分别加212.9 μg/mL没食子酸对照品储备液850 μL,按照“2.2.3”项下方法制备供试品溶液,按“2.2.1”项下条件检测,回收率结果见表1。
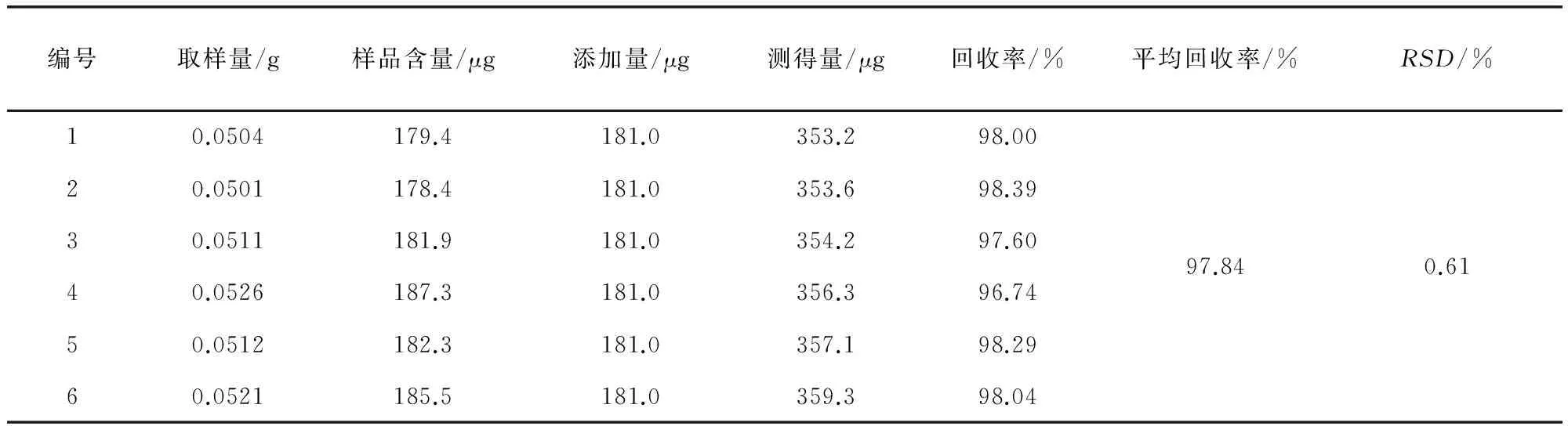
表1 回收率试验结果
2.2.10 样品的测定 取供试品溶液注入液相色谱仪,记录色谱图,按外标法计算没食子酸含量,结果见表2。
3 讨论与小结
3.1 TLC鉴别中展开系统以及薄层板的选择 考察了乙酸乙酯-丁酮-甲酸-水(10∶1∶1∶1)、甲苯(用水饱和)-乙酸乙酯-甲酸(6∶3∶1)、石油醚(60~90℃)(用水饱和)-乙酸乙酯-甲酸(6∶5∶1)以及环己烷(用水饱和)-乙酸乙酯-甲酸(6∶5∶1)4种展开系统,结果表明:采用乙酸乙酯-丁

表2 大黄末中没食子酸含量测定结果
酮-甲酸-水(10:1:1:1)色谱系统分离度较差;采用甲苯(用水饱和)-乙酸乙酯-甲酸(6∶3∶1)Rf值为0.16,偏小;采用石油醚(60℃~90℃)(用水饱和)-乙酸乙酯-甲酸(6∶5∶1)以及环己烷(用水饱和)-乙酸乙酯-甲酸(6∶5∶1)分离度均较好,Rf值分别为0.57和0.60,但前者的斑点有扩散,综合考虑采用环己烷(用水饱和)-乙酸乙酯-甲酸(6∶5∶1)作为展开系统。
对硅胶G薄层板及聚酰胺薄膜做了比较,采用5 μL点样,喷以1%三氯化铁乙醇溶液以后观察,硅胶G薄层板显色更清晰,因此选用硅胶G薄层板。
3.2 含量测定中流动相及超声条件的选择 采用DAD在200 nm~400 nm进行全波长扫描,没食子酸对照品,在215.6 nm与271.0 nm波长处有最大吸收,215.6 nm波长太短,试剂容易产生干扰,故选择271 nm波长作为检测波长。参考文献[4-6]对流动相的组成及不同比例进行了考察,发现流动相中加入磷酸有助于改善峰型,减少拖尾。选择甲醇-0.1%磷酸溶液(15∶85)、甲醇-0.05%磷酸溶液(5∶95)、乙腈-0.05%磷酸溶液(5∶95)等系统作为流动相,试验结果发现,采用甲醇-0.05%磷酸溶液(5∶95)为流动相,没食子酸与其他组分色谱峰可以得到较好的分离。
分别选择超声、加热回流、浸渍30 min的方法考察,结果表明超声与回流提取效果相当,均好于浸渍,考虑到超声操作便捷,故采用超声方法。考察水、25%甲醇、50%甲醇、75%甲醇超声提取30 min,结果表明水的提取效果最佳,这与范晓红等的研究[7]基本一致。
超声15、30、45 min,考察超声时间对试验的影响,结果表明提取时间在15 min时提取不够充分,而当提取时间延长至45 min时与30 min并无显著差异。故选择超声时间为30 min。
本试验所采用的大黄末来自2个兽药企业7批次样品,不同批次大黄末样品中没食子酸的含量存在一定差异。但因采用的大黄药材受炮制加工[8]等因素影响较大,要制订含量限度尚需进行收集大量样品进一步研究。
结果表明,在该薄层色谱条件下,没食子酸的薄层色谱斑点清晰,分离度好;在该液相色谱条件下,没食子酸峰与相邻组分峰的分离度好,该方法的精密度、重复性、回收率试验均符合规定,操作简便、快速,可作为制定大黄末质量标准的参考依据。
[1] 中国兽药典委员会. 中国兽药典二O一O年版二部[S].[2] 中国兽药典委员会. 兽药使用指南中药卷二O一O年版 [S].[3] 匡海学. 中药化学[M]. 北京: 中国中医药出版社, 2003:94.
[4] 王云, 李丽, 张村, 等. 大黄5种饮片没食子酸和儿茶素的含量比较影响[J]. 中国中药杂志, 2010, 35 (17):2267-2269.
[5] 沈华, 王正, 李蕾, 等. HPLC法同时测定没食子提取物中没食子酸、没食子酸甲酯和没食子酸乙酯的含量[J]. 齐鲁药事, 2014, 33(11):631-632.
[6] 张朔生. HPLC测定炮制前后石榴皮中没食子酸的含量[J]. 药物分析杂志, 2010, 30(6):1104-1106.
[7] 范晓红, 李治建, 斯拉甫﹒艾白, 等. 没食子中没食子酸含量测定及总鞣质提取方法的研究[J]. 时珍国医国药, 2011, 22(5):1119-1121.
[8] 雷鹏, 李新中, 朱诗塔, 等. 不同炮制方法对大黄中没食子酸含量的影响[J]. 中药新药与临床药理, 2008, 19(6):477-479.
(编辑:陈希)
Study on the Determination and Identified of Gallic Acid in Rhubarb Powder
Cai Wen-jin, Bao Ai-qing, Lu Chun-bo
(ZhejiangProvinceInstituteofveterinaryDrugandFeedstuff,Hangzhou310012,China)
To perfect the quality control method of rhubarb powder. Gallic acid was identified by thin layer chromatography (TLC) and determined by high performance liquid chromatography (HPLC). A chromatographic column of Agilent Eclipse XDB-C18(250 mm ×4.6 mm, 5 μm) was used, with the mobile phase of methanol - 0.05% phosphoric acid solution (5∶95) at the flow rate of 1.0 mL/min. The detection wavelength was at 271 nm. The calibration curves were linear in ranges of 1.064~106.4 μg/mL(r= 1.0000)for gallic acid; the average recoveries (n=6) was 97.84%(RSD=0.61%). The method is accurate and sensitive for the quality control of rhubarb powder.
rhubarb powder; gallic acid; TLC; HPLC
蔡文金,女,助理兽医师,从事中兽药检验和质量标准研究工作。E-mail:lucb02@163.com
2015-09-06
A
1002-1280 (2016) 01-0052-04
S853.7
猜你喜欢
核化学与放射化学(2022年2期)2022-04-28
昆明医科大学学报(2021年8期)2021-08-13
中成药(2018年6期)2018-07-11
钻井液与完井液(2018年2期)2018-06-13
中国蜂业(2018年4期)2018-05-09
中成药(2017年8期)2017-11-22
中成药(2017年3期)2017-05-17
海峡科技与产业(2016年3期)2016-05-17
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
中国当代医药(2015年8期)2015-03-01
