
氘代Rigosertib类似物的合成*
2016-01-17 08:58胥珂馨陈元伟四川大学生物治疗国家重点实验室四川成都6004成都海创药业有限公司四川成都6004
合成化学 2015年6期
陈 锞,龚 瑜,樊 磊,胥珂馨,陈元伟,(.四川大学生物治疗国家重点实验室,四川成都 6004; .成都海创药业有限公司,四川成都 6004)
氘代Rigosertib类似物的合成*
陈锞1,龚瑜2,樊磊2,胥珂馨2,陈元伟1,2
(1.四川大学生物治疗国家重点实验室,四川成都610041; 2.成都海创药业有限公司,四川成都610041)
摘要:以2,4,6-三羟基苯甲醛为原料制得中间体2,4,6-三甲氧基苯甲醛(2a)和2,4,6-三三氘甲氧基苯甲醛(2b);以4-烷氧基-3-硝基苄硫基乙酸为原料,经氧化、缩合-脱羧、还原、取代和水解5步反应合成了氘代Rigosertib类似物,收率80%~86%,其结构经1H NMR和MS表征。
关键词:2,4,6-三羟基苯甲醛;氘代药物;合成
对先导药物潜在代谢点进行改造,可减慢其在人体内的代谢,达到增强药效的目的。该方法投入小,研发周期短,风险较低,是新药研发的有效思路[1]。碳氘键较碳氢键更稳定[2-3],将氢替换为氘后,既能最大限度的保留药物分子的原有结构,维持其药理活性,又可能增强药物稳定性,延长其半衰期,从而提高药效,减短药物服用周期[4]。
Rigosertib【2-【(2-甲氧基)-5-{[(E)-2-(2,4,6-三甲氧基苯基)乙烯基磺酰基]甲基}苯基】氨基乙酸(Chart 1)】是PLK1的非ATP竞争性抑制剂[5],其对骨髓增生异常综合征患者的治疗已进入三期临床实验阶段。

Chart 1
为进一步提高Rigoserti药理活性,增强其代谢稳定性,我们通过分析Rigosertib结构,找到其潜在代谢位点并进行了氘代改造,合成了6个氘代Rigosertib类似物(7a~7c和8a~8c,Scheme 1)。该反应路线较文献[6]方法收率高,条件温和,操作简便。
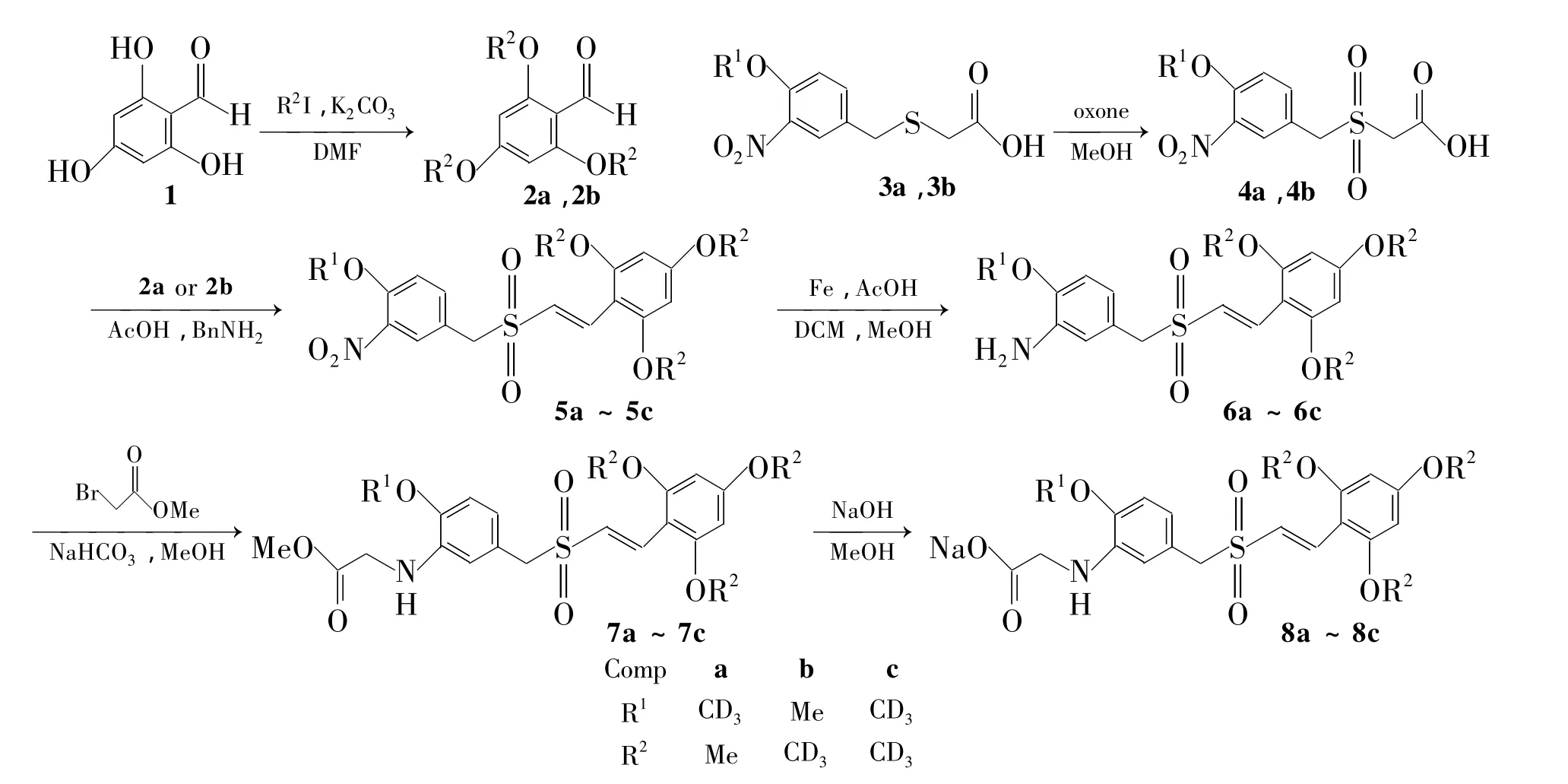
Scheme 1
1 实验部分
1.1仪器与试剂
Bruker AV 400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标); LC-MS 12200-6120型质谱仪[Waters X Bridge C18柱(50 mm×4.6 mm×3.5 μm)]; VARIAN Spectr AA 220FS型原子吸收光谱仪。
4-取代-3-硝基苄硫基乙酸(3a和3b)按文献[6]方法合成;其余所用试剂均为分析纯。
1.2合成
(1)2a和2b的合成(以2a为例)
在反应瓶中加入2,4,6-三羟基苯甲醛(1)1.54 g(10 mmol)和碳酸钾7.0 g的DMF(15 mL)溶液,搅拌下滴加碘甲烷2.5 mL,滴毕,于60℃反应过夜。冷却至室温,加水100 mL,用乙酸乙酯萃取,合并有机相,旋干,剩余物经硅胶柱层析[梯度洗脱剂:V(石油醚)∶V(乙酸乙酯)=50∶1~5∶1]纯化得1.14 g黄色固体2a,收率58%。
以氘代碘甲烷替代碘甲烷,用类似的方法合成2b,收率48%,m.p.121℃~122℃;1H NMR δ:10.36(s,1H),6.07(s,2H)。
(2)4a和4b的合成(以4a为例)
在反应瓶中加入3a 5.1 g(19.6 mmol)和甲醇150 mL,搅拌使其溶解;缓慢滴加过氧硫酸钾26.5 g的水(80 mL)溶液,滴毕,于室温反应10 h。过滤,滤液倒入500 mL水中,用乙酸乙酯萃取,有机层依次用水(100 mL)和饱和食盐水(80 mL)洗涤,用无水硫酸钠干燥,过滤,滤液减压浓缩得黄绿色固体3-硝基-4-三氘代甲氧基苄基磺酰基乙酸(4a)5.6 g,收率98%,m.p.136℃~138℃;1H NMR δ:7.98(s,1H),7.70(d,J=8.7 Hz,1H),7.15(d,J=8.7 Hz,1H),4.53(s,2H),3.86(s,2H); MS m/z:310.3{[M +NH3]+}。
以3b替代3a,用类似的方法合成4b。
(3)5a~5c的合成(以5a为例)
在反应瓶中加入4a 2.0 g(6.8 mmol),2a 1.4 g(7.2 mmol),冰乙酸15 mL和苄胺100 μL,回流反应7 h。冷却至室温,过滤,滤饼用异丙醇洗涤,干燥得粗品,粗品用异丙醇重结晶得黄绿色固体(E)-1,3,5-三甲氧基-2-[2-(4-三氘代甲氧基-3-硝基苄基磺酰基)乙烯基]苯(5a)300 mg,收率11%,m.p.185℃~188℃;1H NMR δ:7.84(d,J=2.0 Hz,1H),7.80(d,J=15.6 Hz,1H),7.63(dd,J=8.7 Hz,2.0 Hz,1H),7.10(d,J=8.7 Hz,1H),7.02(d,J=15.6 Hz,1H),6.09(s,2H),4.22(s,2H),3.85(s,3H),3.84(s,6H); MS m/z:427.2{[M +H]+}。
用类似的方法合成5b和5c。
(4)6a~6c的合成(以6a为例)
在反应瓶中加入5a 300 mg(0.7 mmol)和混合溶剂[V(二氯甲烷)∶V(甲醇)=3∶1]40 mL,搅拌使其溶解;加热至回流,滴加醋酸8 mL,滴毕,缓慢加入铁粉1.3 g,反应8 h。冷却至室温,用硅藻土滤除铁粉,滤液旋蒸除溶,加水析出固体,过滤,滤饼干燥得(E)-2-三氘代甲氧基-5-[(2,4,6-三甲氧基苯乙烯基磺酰基)甲基]苯胺(6a)180 mg; MS m/z:397.2{[M + H]+}。
用类似的方法合成6b和6c。6a~6c均无需纯化,直接投入下一步反应。
(5)7a~7c的合成(以7a为例)
在反应瓶中加入乙酸钾600 mg(39 mmol)和甲醇20 mL,搅拌使其溶解;加入2-溴乙酸甲酯700 μL和6a 160 mg(0.39 mmol),回流反应6 h。除去溶剂,残余物倒入冰水中,用二氯甲烷萃取,合并有机相,用无水硫酸钠干燥。旋干溶剂,剩余物经硅胶柱层析[梯度洗脱剂:V(石油醚)∶V(乙酸乙酯)=30∶1~4∶1]纯化后用乙醇重结晶得白色固体(E)-2-{ 2-三氘甲氧基-5-[(2,4,6-三甲氧基苯乙烯基磺酰基)甲基]苯胺基}乙酸甲酯(7a)101 mg,收率55%,m.p.186℃~188℃;1H NMR δ:7.64(d,J=15.7 Hz,1H),7.00(d,J=15.7 Hz,1H),6.82(d,J=8.1 Hz,1H),6.68(dd,J=8.1 Hz,1.8 Hz,1H),6.47(d,J=1.8 Hz,1H),6.24(s,2H),4.23(s,2H),3.87(d,J=3.3 Hz,12H),3.84(s,2H); MS m/z:469.2{[M + H]+}。
用类似的方法合成7b和7c。
(6)8a~8c的合成(以8a为例)
在反应瓶中加入7a 60 mg(0.128 mmol),甲醇3 mL和二氯甲烷3 mL,搅拌使其溶解;加入10 mol·L-1氢氧化钠溶液500 μL,反应2 h。旋除二氯甲烷,剩余物用HPLC纯化得白色固体8a 50 mg,收率82%,m.p.203℃~205℃;1H NMR δ:7.12(d,J=15.7 Hz,1H),6.80(d,J=15.7 Hz,1H),6.64(d,J=8.0 Hz,1H),6.52(d,J=8.0 Hz,1H),6.29(s,1H),5.85(s,2H),4.13(s,2H),3.63(s,3H),3.55(s,6H),3.26(s,2H); MS m/z:455.2{[M-Na + H]+} ;钠原子吸收含量(×10-6):Calcd 5.17,found 5.48。
用类似的方法合成白色固体8b和8c。
8b:收率80%,m.p.203℃~204℃;
1H NMR δ:7.12(d,J=15.7 Hz,1H),6.80(d,J=15.7 Hz,1H),6.64(d,J=8.0 Hz,1H),6.52(d,J=8.0 Hz,1H),6.29(s,1H),5.83(s,2H),4.12(s,2H),3.64(s,3H),3.26(s,2H); MS m/z:416.2{[M-Na + H]+} ;钠原子吸收含量(×10-6):Calcd 5.56,found 6.33。
8c:收率86%,m.p.203℃~204℃;
1H NMR δ:7.12(d,J=15.7 Hz,1H),6.80(d,J=15.7 Hz,1H),6.64(d,J=8.0 Hz,1H),6.52(d,J=8.0 Hz,1H),6.29(s,1H),5.83(s,2H),4.12(s,2H),3.26(s,2H); MS m/z:464.2{[M-Na +H]+};钠原子吸收含量(×10-6):Calcd 5.19,found 5.68。
2 结果与讨论
合成5a时,由于4a在极性溶剂中的溶解度很小,反应速度慢,反应程度低,加入二氯甲烷后,反应速度加快,反应比较完全。
合成7a时,在柱层析后重结晶,7a纯度大大提高,降低了下一步水解反应后的纯化难度。
合成8a时,由于合成量少,用传统的成盐和重结晶方法不易得到高纯度的8a。我们推测将7a的氢氧化钠水解溶液经低温浓缩后直接用纯水和乙腈进行HPLC制备,可使8a保持羧酸钠盐离子状态。经检测,8a水溶性良好,pH≈9,无无机盐存在,说明8a是羧酸钠盐。另据钠原子吸收含量测定显示,8a分子中仅含一个钠原子,进一步说明8a是以羧酸钠盐的形式存在。
参考文献
[1]Camille G W.The Practice of Medicinal Chemistry [M].Academic Press,2008.
[2]Westheimer F H.The magnitude of the primary kinetic isotope effect for compounds of hydrogen and deuterium [J].Chem Rev,1961,61(3):265-273.
[3]Robert F,Gautier D,Dubrulle B.The solar system D/H ratio:Observations and theories[J].Space Sci Rev,2000,92:201-224.
[4]Thomas G.Gant,using deuterium in drug discovery:Leaving the label in the drug[J].J Med Chem,2014,57(9):3595-3611.
[5]杨臻峥.抗肿瘤药ON.01910.Na[J].药学进展,2012,36(1):45-46.
[6]M V R Reddy,P Venkatapuram,Muralidhar R,et al.Discovery of a clinical stage multi-kinase inhibitor sodium(E)-2-{2-methoxy-5-[(2',4',6'-trimethoxystyrylsulfonyl)methyl]phenylamino}-acetate(ON.01910.Na):Synthesis,structure activity relationship and biological activity[J].J Med Chem,2011,54:6254-6276.
·制药技术·
Synthesis of Deuterated Rigosertib Analogs
CHEN Ke1,GONG Yu2,FAN Lei2,XU Ke-xin2,CHEN Yuan-wei1,2
(1.State Key Laboratory of Biotherapy,Sichuan University,Chengdu 610074,China; 2.Hinova Pharmaceuticals Inc.,Chengdu 610074,China)
Abstract:Two intermediates,2,4,6-trimethoxybenzaldehyde(2a)and 2,4,6-trideuteromethoxybenzaldehyde(2b),were prepared from 2,4,6-trihydroxybenzaldehyde.Three deuterated Rigosertib analogs,in yield of 80%~86%,were synthesized by the reaction of oxidization,condensation-decarboxylation,reduction,substitution and hydrolysis from[(4-alkaxy-3-nitrobenyl)-thion]acetic acid.The structures were confirmed by1H NMR and MS.
Keywords:2,4,6-trihydroxybenzaldehyde; deuterated drug; synthesis
通讯作者:陈元伟,教授,E-mail:ywchen@ hinovapharma.com
作者简介:陈锞(1985-),男,汉族,重庆綦江人,硕士研究生,主要从事有机合成的研究。E-mail:chenke006@163.com
收稿日期:2014-08-25;
修订日期:2015-04-27
DOI:10.15952/j.cnki.cjsc.1005-1511.2015.06.0557 *
文献标识码:A
中图分类号:O625.32; R914.5
猜你喜欢
肝博士(2022年3期)2022-06-30
化学工程师(2022年3期)2022-04-19
上海化工(2021年2期)2021-04-23
天然产物研究与开发(2019年1期)2019-03-01
中国洗涤用品工业(2017年2期)2017-04-16
当代化工研究(2016年2期)2016-03-20
中国洗涤用品工业(2016年2期)2016-02-28
科技与企业(2015年20期)2015-10-21
医学研究杂志(2015年6期)2015-07-01
应用化工(2014年5期)2014-08-08
