
超高效液相色谱-四极杆-飞行时间质谱法快速筛查茶叶中的204 种农药残留
2015-12-26 01:57吕亚宁赵暮雨周芳芳胡艳云
色谱 2015年6期
余 璐, 宋 伟, 吕亚宁, 赵暮雨,周芳芳, 胡艳云, 郑 平*
(1. 安徽农业大学茶与食品科技学院,安徽 合肥230036;2. 安徽出入境检验检疫局,安徽 合肥230022;3. 食品安全分析与检测安徽省重点实验室,安徽 合肥230022)
茶叶中的农药残留问题一直是各国关注的焦点。许多国家及国际组织均对茶叶中的农药残留制定了最大残留限量(MRLs),例如,日本肯定列表制度规定与茶叶有关的农药种类有276 种,欧盟有453 种,中国有28 种[1-3]。随着人们对食品安全的日益重视,各国规定的检测项目还在不断增加。因此,迫切需要发展一种高通量的农药残留快速检测技术。
目前,气相色谱-四极杆质谱法(GC-Q/MS)和液相色谱-三重四极杆串联质谱法(LC-QQQ/MS)由于选择性好、灵敏度高而广泛应用于茶叶中的农药多残留分析[4-8]。但以四极杆作为质量分析器的GC-Q/MS 和LC-QQQ/MS 均为低分辨质谱,当分析复杂样品时,往往对质荷比接近的干扰物不能有效地区分,常出现假阳性结果。同时在选择离子扫描(SIM)或多反应监测(MRM)扫描模式下,由于仪器扫描速率不高导致离子驻留时间有限,限制了一次同时扫描的化合物数量,无法真正实现高通量筛查。在采用LC-QQQ/MS 分析几百种农药时,往往需要把化合物分成几组进行分别检测[9-11],降低了分析速度。而在全扫描模式时,其灵敏度低,分辨率差,不能准确定性[12]。此外,GC-Q/MS 和LCQQQ/MS 的定性分析必须依赖标准物质。而标准物质的配制和使用,增加了实验成本和分析的工作量。近年来高分辨质谱的应用为农药多残留分析提供了可靠的依据。
高分辨的飞行时间质谱(TOF/MS)具有质量范围广、分辨率和质量精度较高、分析速度快的特点。与低分辨质谱不同,TOF/MS 可通过全扫描获得化合物的精确质量数和可能的化学分子式,大大提高了复杂背景下的抗干扰能力,使检测结果更加准确可靠。并且TOF/MS 的扫描速率高,理论上同时扫描的目标物数量无上限,可真正实现一次扫描几百种农药的高通量检测[13-15]。TOF/MS 还可建立特定化合物数据库,结合样品采集的精确质量数、保留时间、同位素比值等信息,通过软件进行自动检索和分析确证,从而实现不使用标准品对目标化合物进行快速鉴定[16,17]。因此,TOF/MS 是对复杂样品中痕量化合物进行定性分析的有效手段,可满足高通量快速筛查和定量分析的需求,在农药多残留检测方面具有良好的应用前景。
本实验以茶叶为研究对象,以固相萃取法为净化手段,采用超高效液相色谱-四极杆-飞行时间质谱(UPLC-Q-TOF/MS)仪,建立了茶叶中204 种农药的快速筛查方法。该方法基于UPLC-Q-TOF/MS技术建立了204 种农药的精确质量数据库和谱图库,利用数据库对质谱检测结果的检索进行筛查分析,从而实现了无需标准品对照,一次进样就可完成茶叶中204 种农药的同时筛查与确证。该方法具有快速、灵敏、准确的特点,为茶叶中农药的高通量快速检测提供了可靠的分析平台。
1 实验部分
1.1 仪器、试剂与材料
Agilent 1290-6540 超高效液相色谱-四极杆-飞行时间质谱仪(美国Agilent 公司);ZORBAX SBC18 柱(100 mm×2.1 mm,3.5 μm)(美国Agilent公司);匀浆机(德国IKA 公司);旋转蒸发仪(瑞士BUCHI 公司);甲酸、甲醇、甲苯和乙腈均为色谱纯(美国TEDIA 公司);石墨化炭黑-N-丙基乙二胺复合固相萃取柱(Carb-PSA,6 mL,500 mg,SUPELCO 公司);无水硫酸钠为分析纯;实验用水均为超纯水(电阻率为18.2 MΩ·cm)。
204 种农药标准品:纯度≥98%,购自德国Dr.Ehrenstorfer 公司。
标准溶液的配制:i)农药单标准溶液:准确称取各农药标准品10 mg(精确至0.01 mg),分别置于10 mL 的棕色容量瓶中,根据其溶解性选择甲醇、乙腈、丙酮等溶剂[2,13,15]溶解并定容至刻度,于4 ℃避光保存。ii)农药混合标准溶液的配制:按照每种农药的化学性质和保留时间把农药分成A、B、C、D 4组。根据单标准溶液的浓度,准确量取适量单标准溶液至100 mL 容量瓶中,用甲醇定容至刻度,于4℃避光保存。
茶叶样品为实验室日常送检样品。
1.2 样品前处理
称取1 g(精确至0.01 g)茶叶于50 mL 离心管中,加入15 mL 乙腈,13 500 r/min 均质提取1 min,4 200 r/min 离心5 min,吸取上层清液于鸡心瓶中。残渣用15 mL 乙腈重复提取一次,离心。合并上清液,40 ℃水浴旋转蒸发至1 mL 左右,待净化。
Carb-PSA 柱中加入约2 cm 高的无水硫酸钠,用4 mL 乙腈-甲苯(3 ∶1,v/v)预洗柱,用下面连接的鸡心瓶收集流出液,将流出液转移至Carb-PSA柱,并用乙腈-甲苯(3 ∶1,v/v)洗涤鸡心瓶3 次(每次2 mL),将洗涤液也移入柱中;在柱上装上50 mL贮液器,用25 mL 乙腈-甲苯(3 ∶1,v/v)淋洗柱,收集所有流出液于鸡心瓶中,在40 ℃水浴中旋转蒸发至0.5 mL,用氮气吹干,用1.5 mL 乙腈-0.1% 甲酸水(2 ∶8,v/v)定容液溶解,过0.2 μm 滤膜,滤液供仪器测定。
1.3 UPLC-Q-TOF/MS 条件
1.3.1 色谱条件
色谱柱:ZORBAX SB-C18柱;流动相:A 相为5 mmol/L 乙酸铵-0.1% (v/v)甲酸水溶液,B 相为乙腈。梯度洗脱程序:0 ~3.00 min,1% B ~30% B;3.00~6.00 min,30% B ~40% B;6.00 ~9.00 min,40% B;9.00 ~15.00 min,40% B ~60% B;15.00 ~19.00 min,60% B ~90% B;19.00 ~23.00 min,90% B;23.00~23.01 min,90% B ~1% B,保持4 min。流速:0.4 mL/min;柱温:40 ℃;进样量:10 μL。
1.3.2 质谱条件
离子源:电喷雾电离(ESI)源,正离子模式;干燥气温度:325 ℃;干燥气流速:10 L/min;雾化器压力:276 kPa;鞘流气温度:325 ℃;鞘流气流速:11 L/min;毛细管电压:4 000 V;扫描方式:正离子全扫描;全扫描范围:m/z 50 ~1 600;碎裂电压:140 V。
UPLC-Q-TOF/MS 配置了双喷雾器电喷雾源,可以连续导入参比溶液对仪器质量轴进行实时校正。参比溶液中含嘌呤(C5H4N4,其离子精确相对分子质量为121.050 873 0)和HP-0921 (C18H18O6N3P3F24,其离子精确相对分子质量为922.009 798),能够实时对测定的目标物进行质量数校正,给出离子的准确质量。
1.4 数据库的建立
1.4.1 一级精确质量数据库的建立(TOF/MS 模式)
本实验在上述色谱分离条件下对204 种农药的标准溶液进行分析,获得204 种农药的保留时间、精确相对分子质量、母离子以及离子化形式。通过在系统自带软件中输入每种农药的名称、分子式、精确相对分子质量和保留时间,建立了204 种农药的一级精确质量数据库。
1.4.2 二级谱图库的建立(Q-TOF/MS 模式)
一级精确质量数据库建立完成后,在不同碰撞能量下对204 种农药进行再次测定,采集二级谱图库所需数据,然后通过PCDL 软件,建立204 种农药的二级谱图库,包括母离子、保留时间、不同碰撞能量下的二级质谱图等信息。
2 结果与讨论
2.1 提取方法的选择
目前,茶叶中农残检测常采用乙腈作为提取试剂。经研究证明,乙腈极性较大,穿透能力强,能提取范围较宽、种类较多的农药样品。且相对其他试剂而言,乙腈提取的色素等杂质较少[4,9,18]。因此,本实验选用乙腈作为提取试剂。
针对茶叶样品前处理,目前日本官方分析方法以及大部分文献[9,19-22]报道采用茶叶加水浸泡,然后采用有机溶剂萃取的方式。而也有少量文献[5,11,23]报道采用直接加入有机试剂均质提取的方式。因此本实验对这两种提取方式进行了考察。结果表明,加水浸泡后,农药的提取效率并没有得到显著改善,提取回收率为51.03% ~153.26%;且随着加水浸泡,茶叶中的一些水溶性色素或水溶性杂质(如茶多酚、茶碱等)也被提取出来,使基质干扰加大,导致噻虫啉、啶虫脒、抑霉唑、双酰草胺、抗蚜威、萎锈灵等农药的响应普遍偏低,降低了碎片离子的匹配程度,从而在筛查过程中出现假阴性现象。本实验采用不加水直接用乙腈提取样品。另外,本实验对均质、手动涡旋和振荡器振荡3 种提取方式进行了比较。结果发现,均质的提取效率最高,对204种农药的提取回收率为64.30% ~122.47%,基质干扰相对较小;提取时间最短,明显优于其他方法;且对204 种农药均能准确定性。这是因为高速均质可以破坏样品组织,使提取试剂与目标物充分接触,从而提高萃取效率。综合考虑各种因素,本实验采用乙腈均质提取的方法。
2.2 净化方法的选择
茶叶基体复杂,含有大量的色素、生物碱、有机酸等,不仅会干扰目标物的分析,而且会对色谱柱和质谱造成致命的损害,故需要有效的净化方法去除杂质干扰。本实验采用乙腈-甲苯(3 ∶1,v/v)作为洗脱液,比较了常用于农残检测的4 种固相萃取小柱:Cleanert TPT 柱、Carb-PSA 柱、Florisil 柱和Envi-Carb 柱。以基质效应较大的7 种农药为例,测试结果见图1。结果发现,Florisil 柱或Envi-Carb 柱的回收率高于Carb-PSA 柱与Cleanert TPT柱。Carb-PSA 柱与Cleanert TPT 柱的回收率结果较为相近,其204 种农药的加标回收率分别在65.2% ~119.5% 与70.3% ~116.4% 范围内。从净化效果看,Florisil 柱去除色素的能力较差;Envi-Carb 柱虽可去除大部分色素,但去除其他干扰物的能力一般,净化效果较差。Carb-PSA 柱是双NH2结构,具有较高的离子交换容量,能够有效去除茶叶中如色素、有机酸等极性杂质,样品提取液谱图上的杂质峰较少,基质效应更小。综合来看,虽然Florisil 柱或Envi-Carb 柱的回收率较高,但大量杂质也同时随之洗脱下来,净化效果较差。而Carb-PSA柱与Cleanert TPT 柱的净化效果较好,且204 种农药的回收率均满足实验要求。另外,考虑到Carb-PSA 柱的使用成本较Cleanert TPT 柱要低,故实验采用Carb-PSA 固相萃取小柱净化。
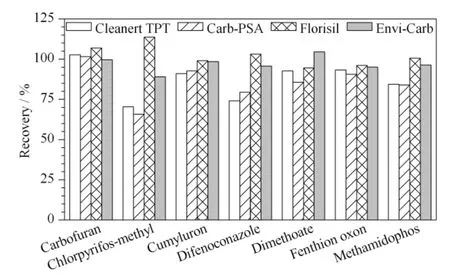
图1 7 种农药在不同固相萃取柱上的回收率对比Fig.1 Recoveries of the seven pesticides on different SPE columns
通过洗脱曲线对洗脱溶剂的用量进行了优化。在空白样品中加入50 μg/kg 混合标准品后进行洗脱,比较了5、10、15、20、25、30 mL 洗脱溶剂对204种农药回收率的影响,部分农药的洗脱曲线见图2。结果表明,当洗脱溶剂体积≤10 mL 时,大部分农药的回收率随着洗脱溶剂体积的增加而增大。当洗脱溶剂体积≥15 mL 时,只有吡蚜酮(pymetrozine)和嘧螨醚(pyrimidifen)的回收率继续增加,其余农药的回收率均趋于平稳。洗脱溶剂体积为25 mL 时,204 种农药的回收率为70.34% ~118.38%,且全部趋于平稳,故选择洗脱溶剂的用量为25 mL。
2.3 定性分析
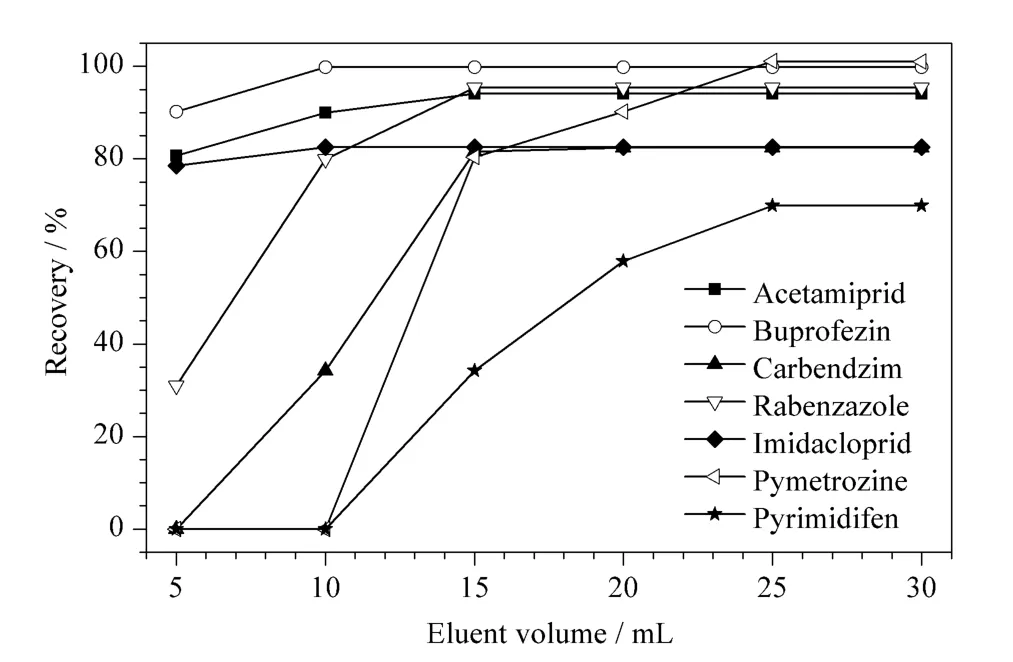
图2 洗脱溶剂用量对部分农药回收率的影响Fig.2 Effect of eluent volume on the recoveries of a part of pesticides
在实际样品分析中,样品先进行一级全扫描测定,测定结果通过已建立的一级精确质量数据库进行自动检索,软件根据实测离子与数据库中的精确质量偏差、保留时间、同位素分布和同位素比例4 个因素的匹配程度进行打分,经优化,将检索得分≥70的农药确定为疑似农药。以茶叶空白样品中添加10 μg/kg 噻嗪酮为例,图3a 为噻嗪酮的一级提取离子流色谱图,图3b 为一级质谱图,图3c 为软件自动检索后噻嗪酮的一级匹配结果。由图3c 可知,噻嗪酮的质量偏差均小于5×10-6(5 ppm),满足欧盟的定性要求,可以作为定性依据。且保留时间(图3a)、同位素比例及其分布(图3b)与数据库中的理论值也均匹配良好,一级检索得分为97.50,从而确定噻嗪酮为疑似农药。由此可见,本实验可以根据精确质量偏差、保留时间、同位素分布和同位素比例对样品中的204 种农药进行快速筛查。
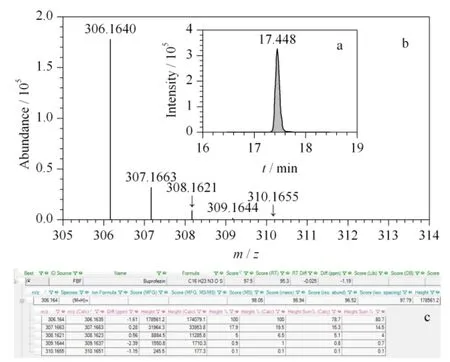
图3 噻嗪酮的(a)提取离子色谱图、(b)一级质谱图和(c)一级匹配结果Fig.3 (a)extracted ion chromatogram,(b)MS spectrum and (c)MS matched results of buprofezin
筛查出的疑似农药还需利用二级质谱的碎片离子进行进一步确证。样品经二次测定后,将样品的碎片离子信息与谱图库中碎片离子信息进行匹配,给出镜像对比结果。经优化,二级检索得分≥60 的农药确定为目标农药。图4 为噻嗪酮的二级质谱镜像对比结果,图4 中可以明显看到主要的特征碎片与谱图库均匹配良好,噻嗪酮的二级得分为94.93,故确定样品中含有噻嗪酮。参照欧盟2002/657/EC[24]规定:当使用高分辨质谱时,每个离子可获得2 个定性位点。而UPLC-Q-TOF/MS 通过全扫描获得一级母离子的精确质量数和二级碎片离子信息,噻嗪酮可获得22 个定性位点,完全满足定性要求(限用化合物确认需最少3 个定性位点),可实现在无对照标准品的情况下进行筛查与确证。此外,噻嗪酮通过UPLC-Q-TOF/MS 获得的定性位点远远高于LC-MS/MS 的4 个位点,从而大大降低了出现假阳性结果的概率,提高了定性结果的可信度。
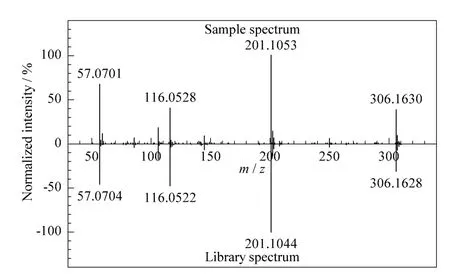
图4 茶叶样品中噻嗪酮的二级质谱镜像结果Fig.4 MS2 spectral difference results of buprofezin in a tea sample
为了考察本实验所建立的筛查方法的可靠性,采用已建立的一级精确质量数据库和二级谱图库对添加了204 种农药的茶叶样品进行自动检索。结果见表1,204 种农药均与一级精确质量数据库检索匹配良好,质量偏差均小于5 ppm,检索得分均高于70。而二级谱图库检索得分高于60 的占91.7% ;少部分农药得分低于60,这可能是由于添加的农药浓度较低或基质导致有相近质量数的干扰造成的,但其谱图中主要的特征离子均匹配良好。在实际样品分析中,这种情况会经常出现,一般通过扣除背景来减少样品基质对定性匹配的干扰。在采用该方法对基质加标样品的分析中,二级检索得分低于60 的农药,扣除背景后得分均高于60,从而确定为目标农药。
由此可见,本实验建立的筛查方法是准确可靠的,能够在无对照标准品的情况下完成茶叶中204种农药的筛查与确证。
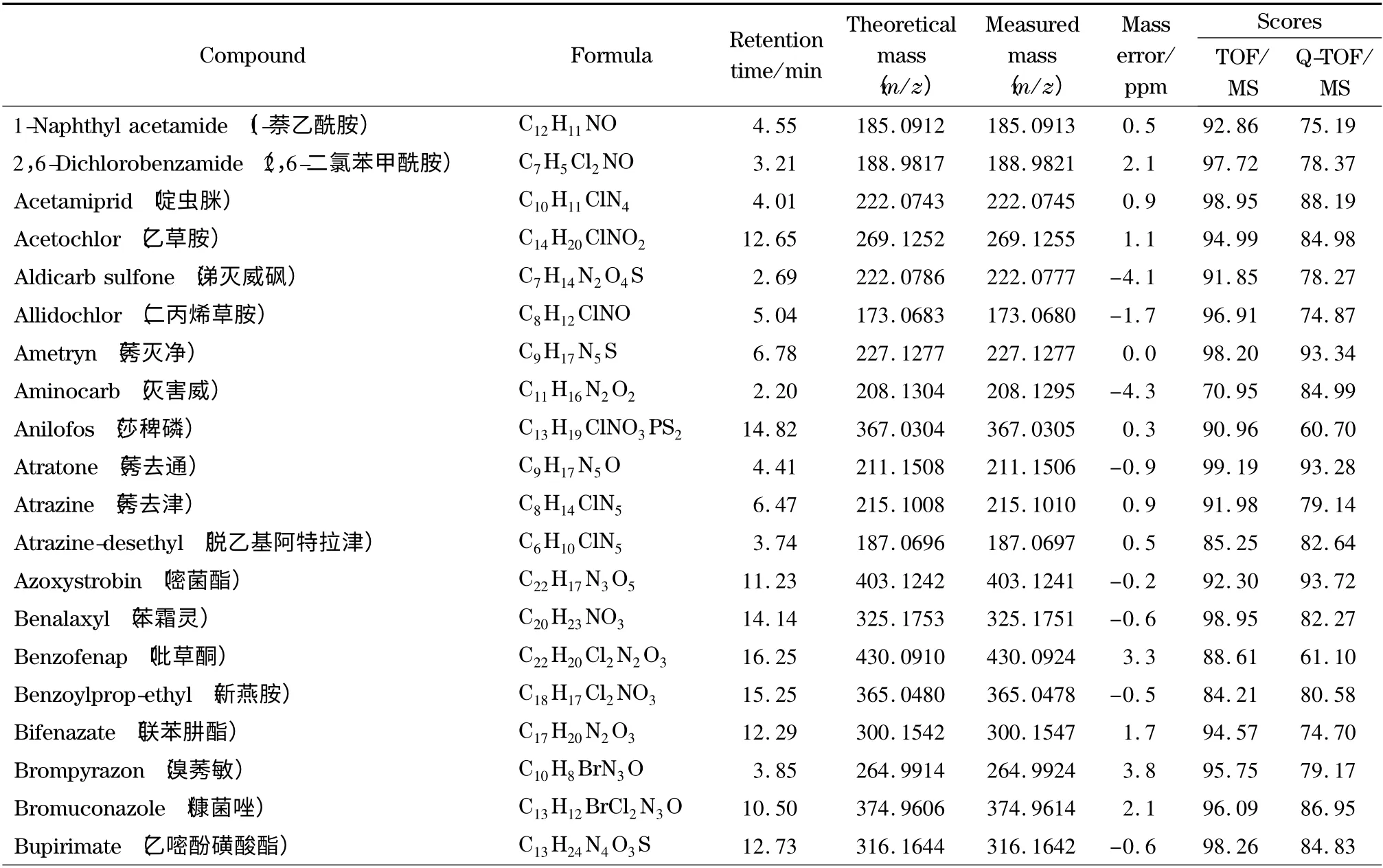
表1 204 种农药的分子式、保留时间、精确质量数、质量数偏差和检索得分Table 1 Formulae,retention times,accurate masses,mass errors and automated retrieval scores of the 204 pesticides
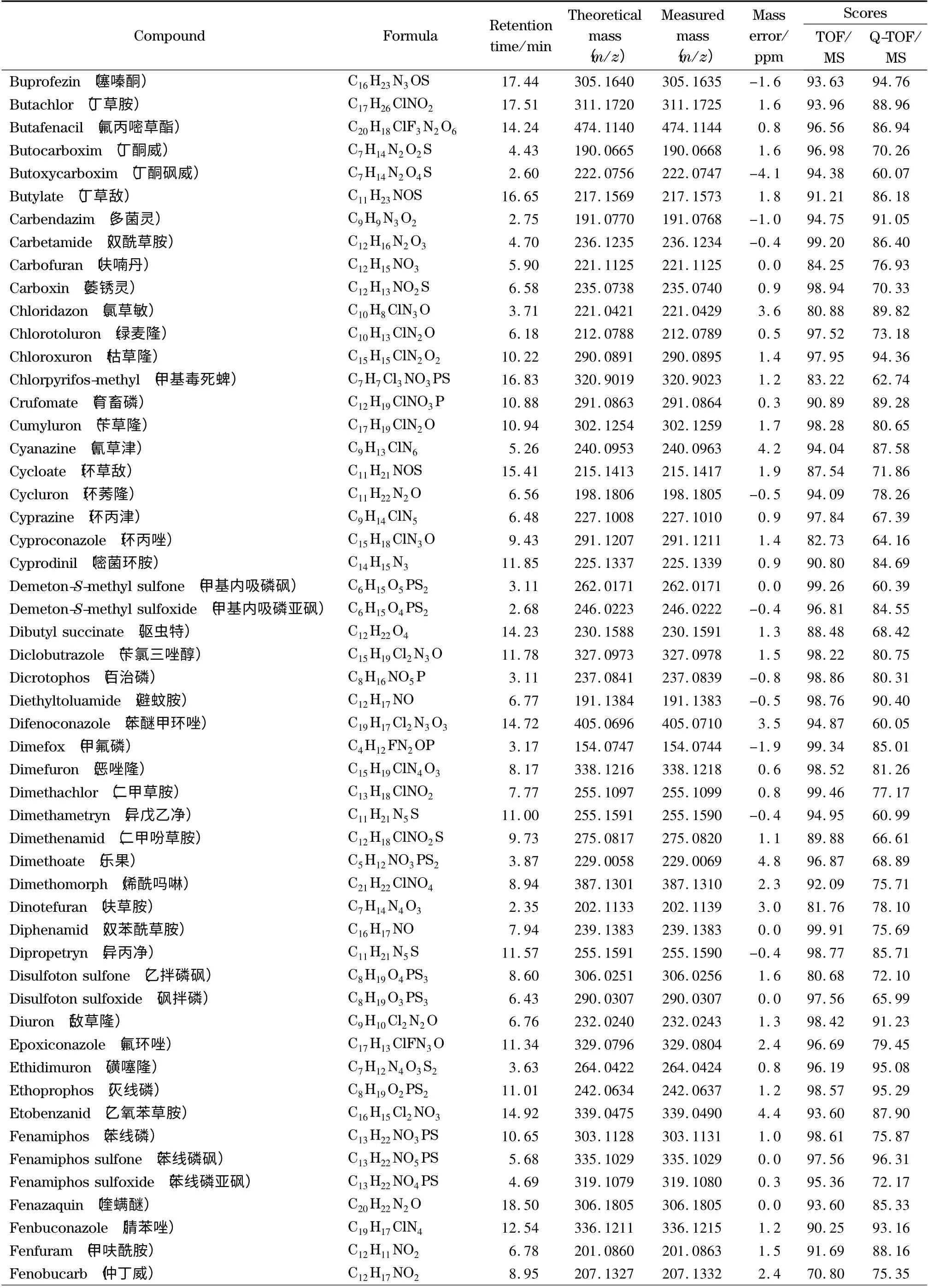
表1 (续)Table 1 (Continued)
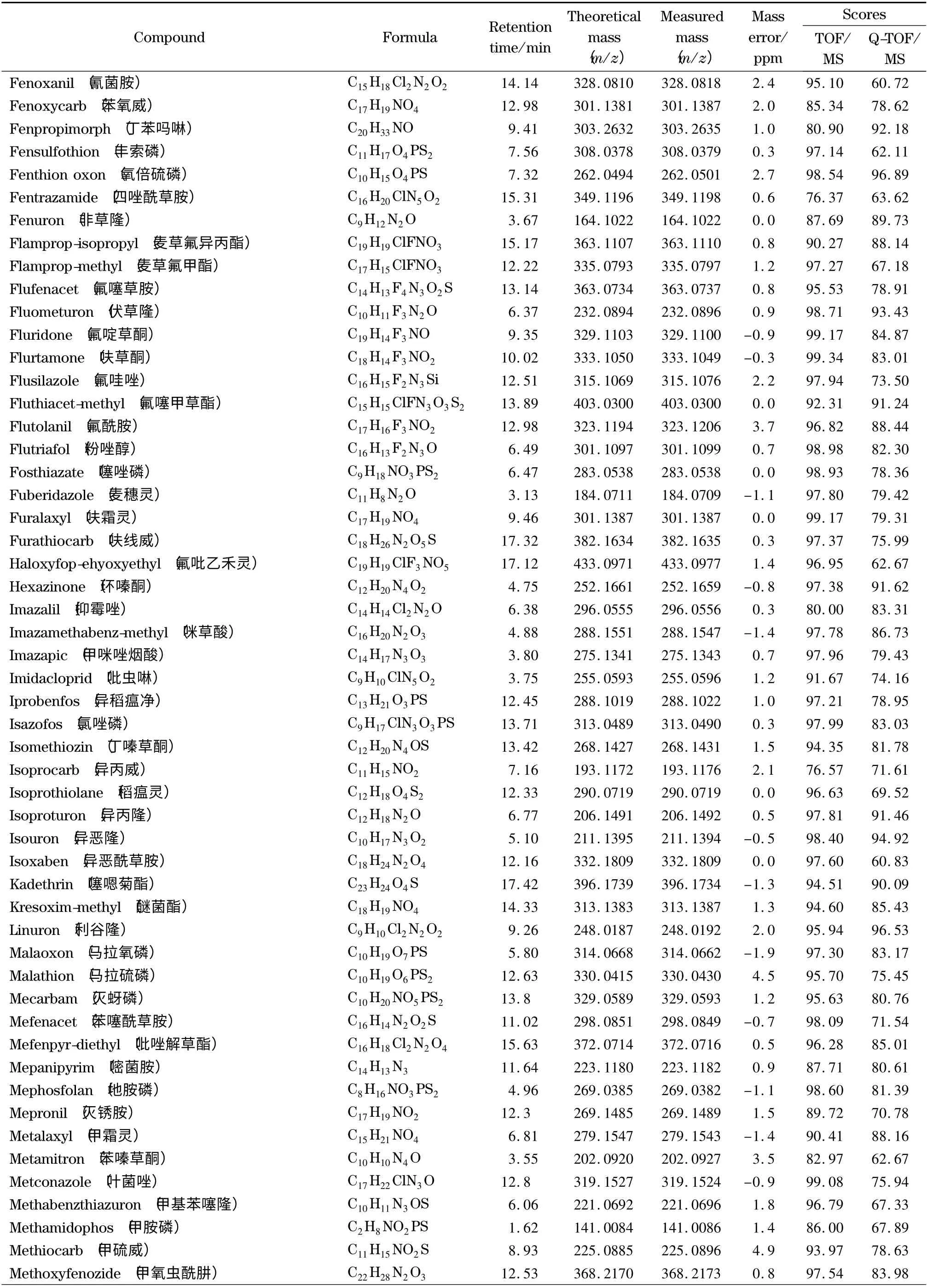
表1 (续)Table 1 (Continued)
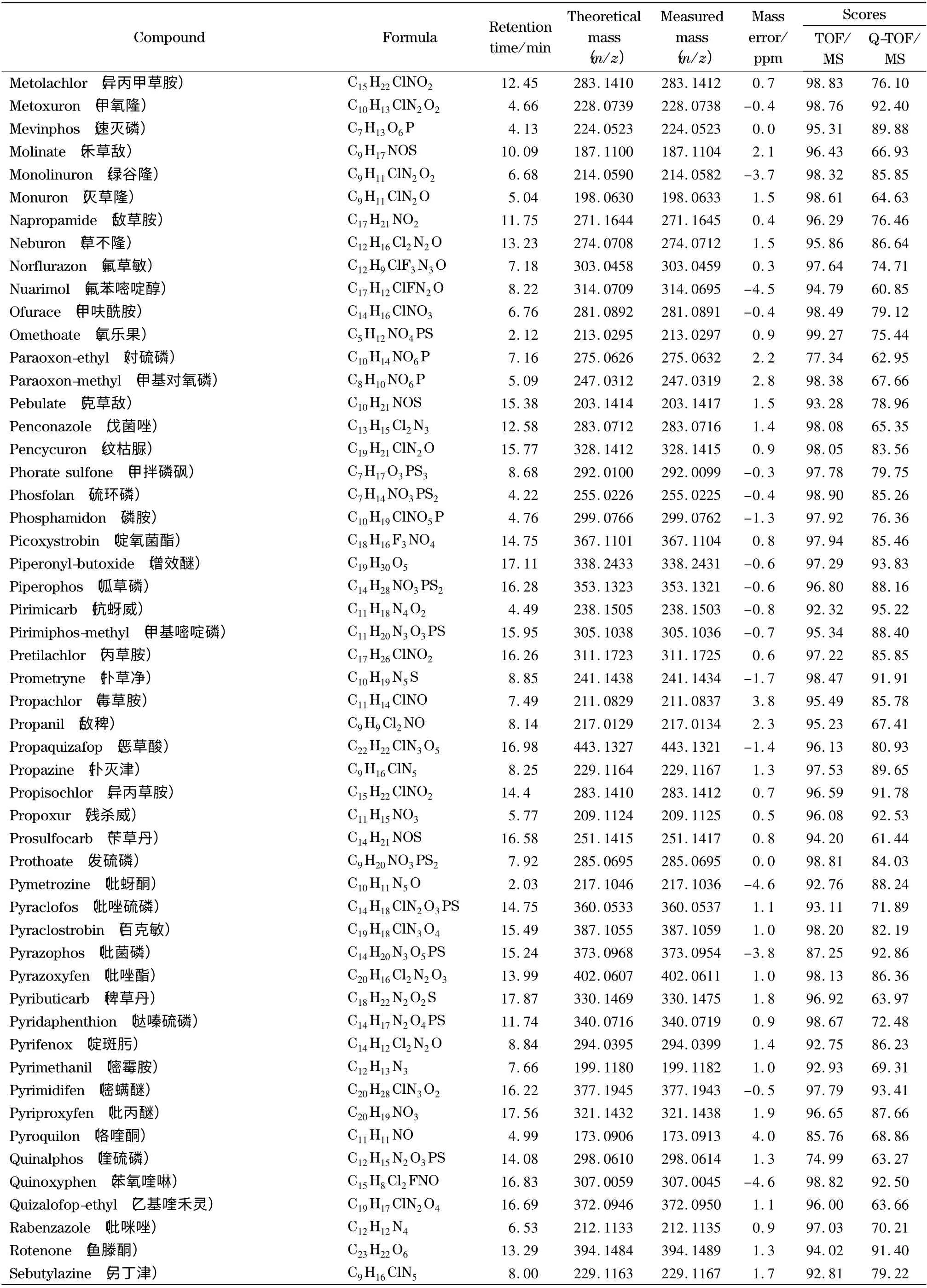
表1 (续)Table 1 (Continued)
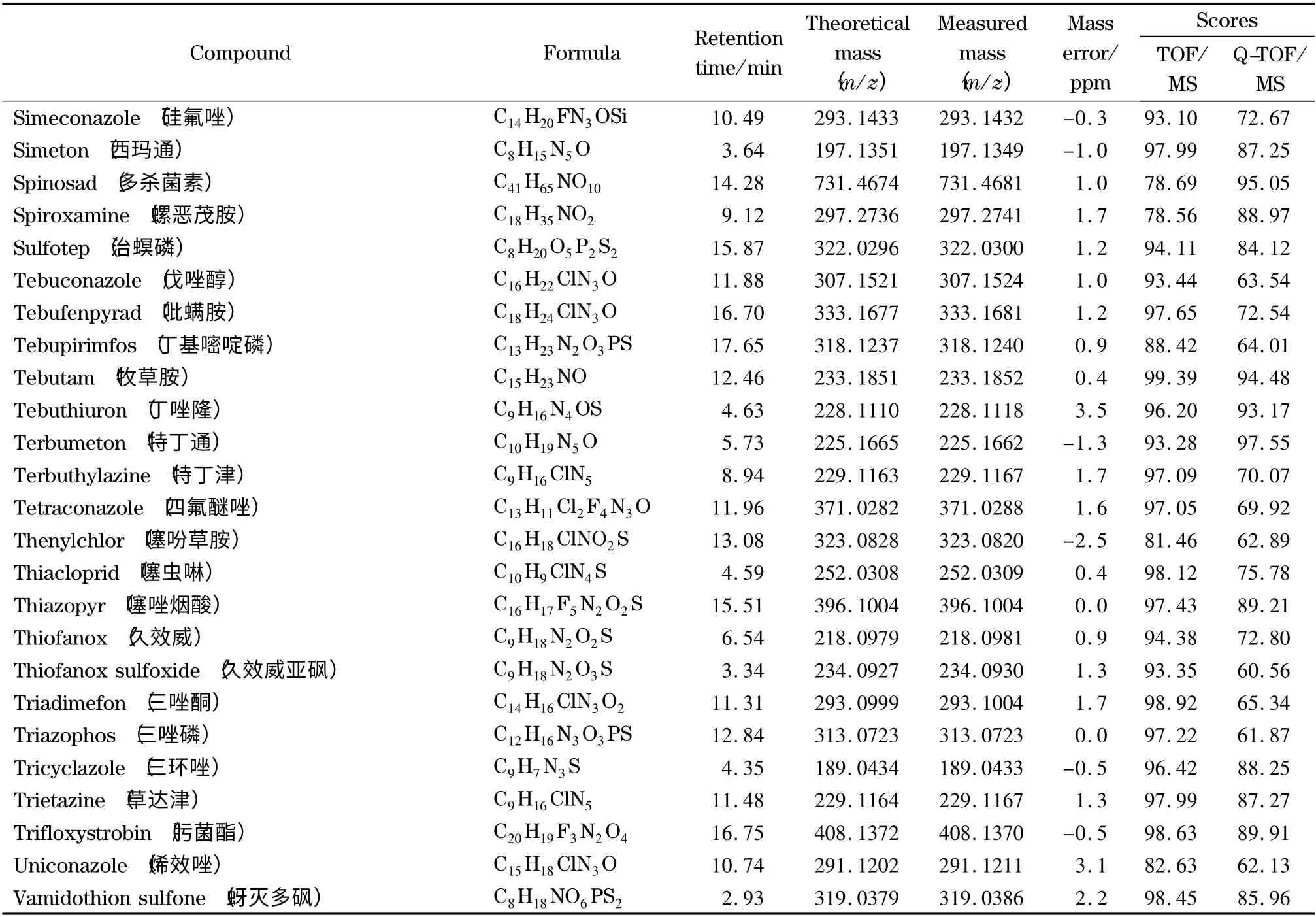
表1 (续)Table 1 (Continued)
2.4 基质效应的评价
基质效应是指样品中除了目标分析物以外的其他成分对待测物测定值的影响,也就是指基质对分析方法准确性的干扰。本文分别用基质空白液和溶剂配制了一系列不同质量浓度(5、10、20、50、100 μg/L)的混合标准溶液,以峰面积对应的质量浓度分别做标准曲线,然后比较这两条标准曲线斜率的差异,从而判断基质效应的强弱。计算公式如下:基质效应(ME)=[(基质匹配标准曲线的斜率/溶剂标准曲线的斜率)-1]×100%。结果如图5 所示,茶叶中只有3 种农药出现了基质增强效应,其余201种农药的ME 值均为负,所以为基质抑制效应。本实验茶叶中204 种农药中有114 种农药表现了中等基质效应,74 种农药表现强基质效应。中等和强基质效应占总数的92.16%,由此可见茶叶具有较强的基质效应。因此,分析目标化合物时选用空白茶叶配制的基质标准曲线进行定量更为准确。
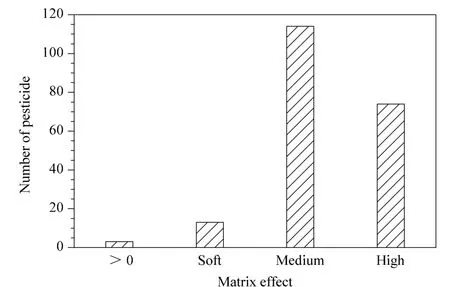
图5 204 种农药在茶叶中的农药基质效应分布Fig.5 Distribution of matrix effects (ME)of the 204 pesticides in tea
目前,基质效应的产生机制还不是十分清楚,在液相色谱-质谱分析中,一般认为基质效应是由质谱检测器的离子化过程引起的。在形成带电喷雾滴时,非挥发性的基质组分与分析物离子竞争产生,非挥发性基质组分将雾滴吸在一起,阻止其分裂成更小的微滴[25]。通常情况下使用电喷雾离子源(EIS)作为电离源时,更容易出现离子化抑制现象,最终导致基质抑制效应。此外,还有很多其他因素也会影响基质效应,如样品基质的种类和浓度,待测物的化学结构和性质等,待测物在样品中的浓度,检测器及接口类型等[26]。
2.5 方法的线性关系、定量限、回收率和精密度
将204 种农药的混合标准溶液用空白样品提取液稀释配制成5、10、20、50、100 μg/kg 的系列基质标准溶液,在建立的分析条件下对其进行测定,以峰面积(y)对质量浓度(x)做标准曲线。由表2 可以看出,204 种农药均线性关系良好,相关系数R2均大于0.99。
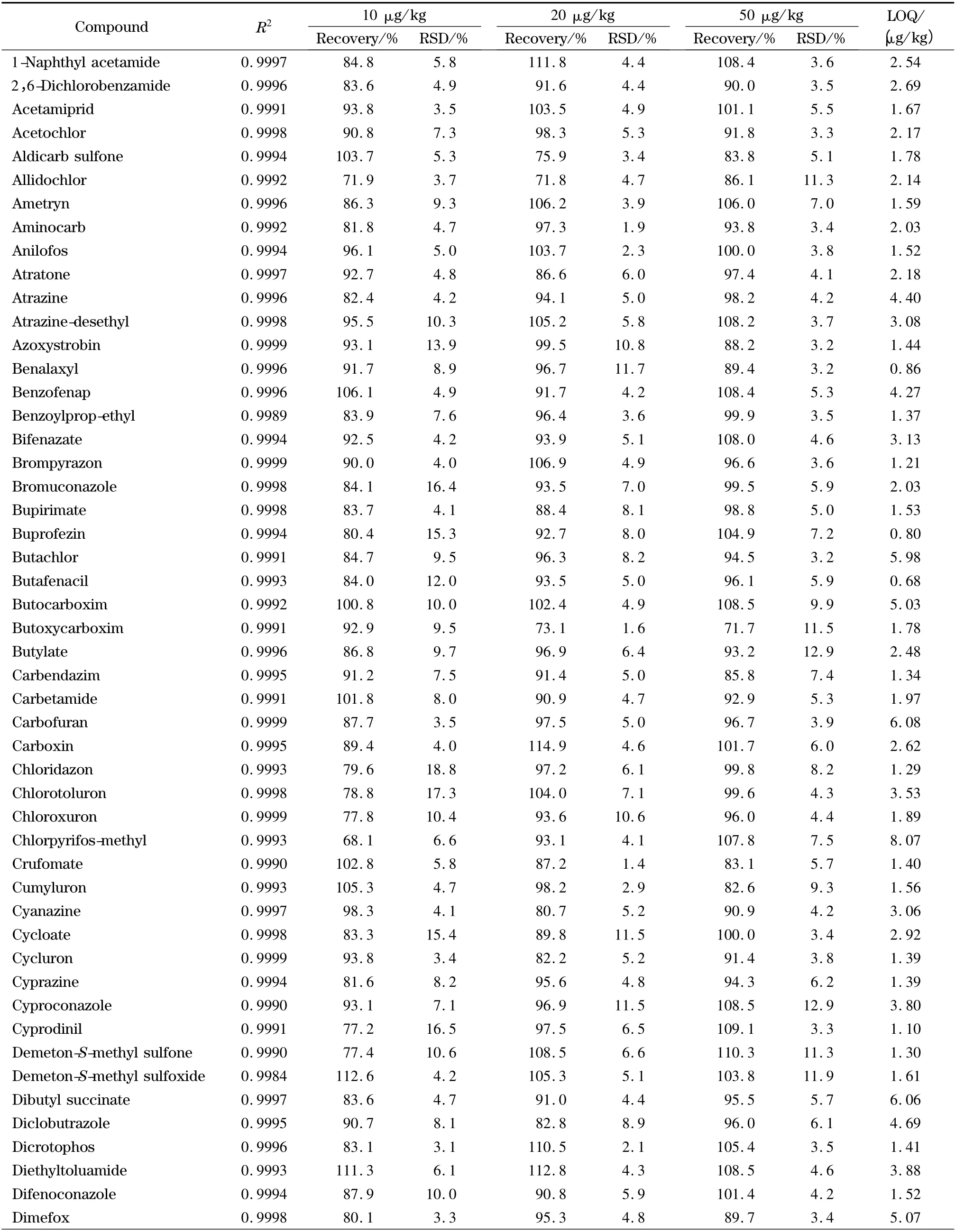
表2 茶叶中204 种农药的相关系数(r2)、添加回收率、相对标准偏差和定量限(n=6)Table 2 Correlation coefficients (R2),spiked recoveries,RSDs and LOQs of the 204 pesticides in tea (n=6)
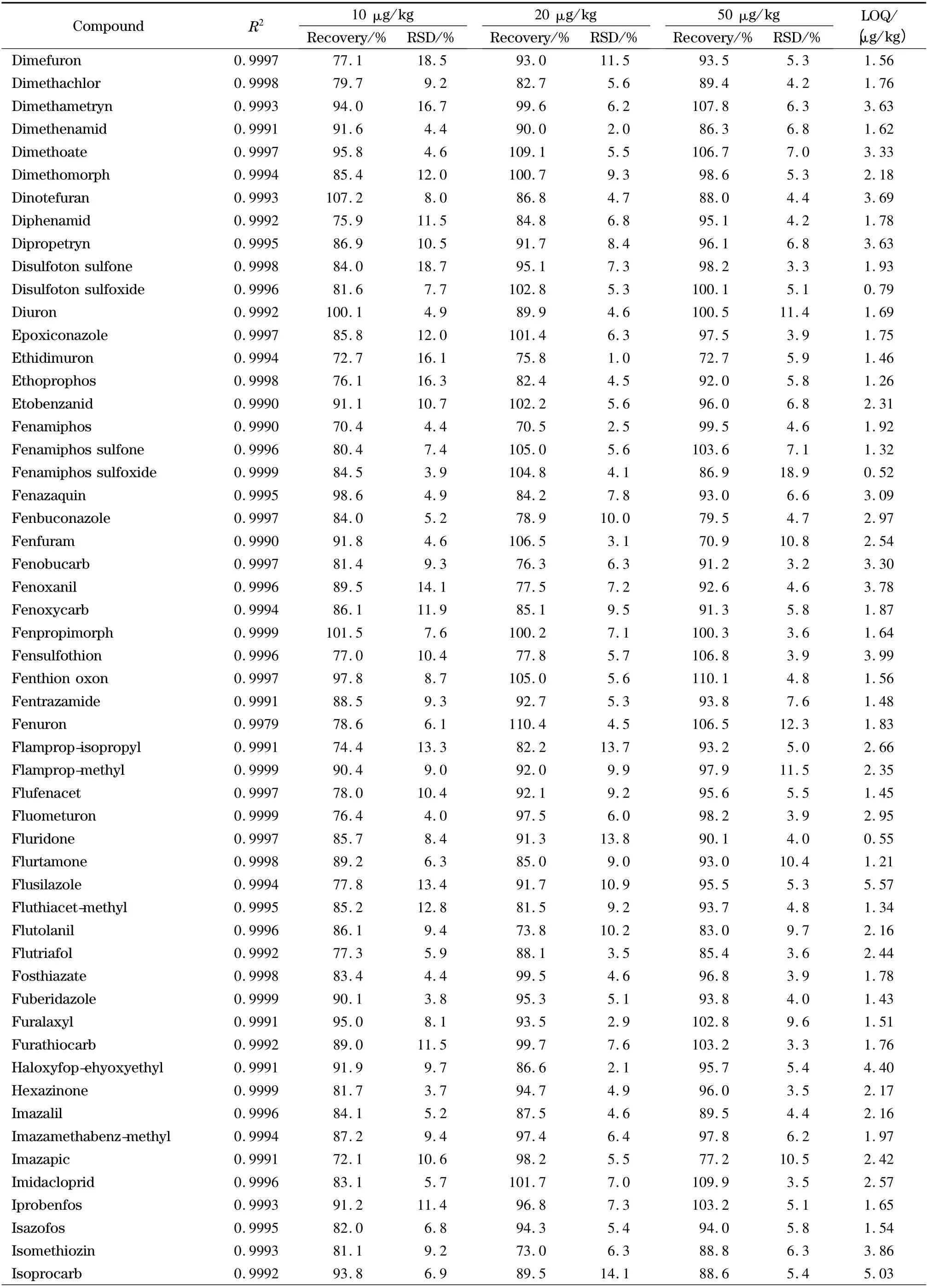
表2 (续)Table 2 (Continued)
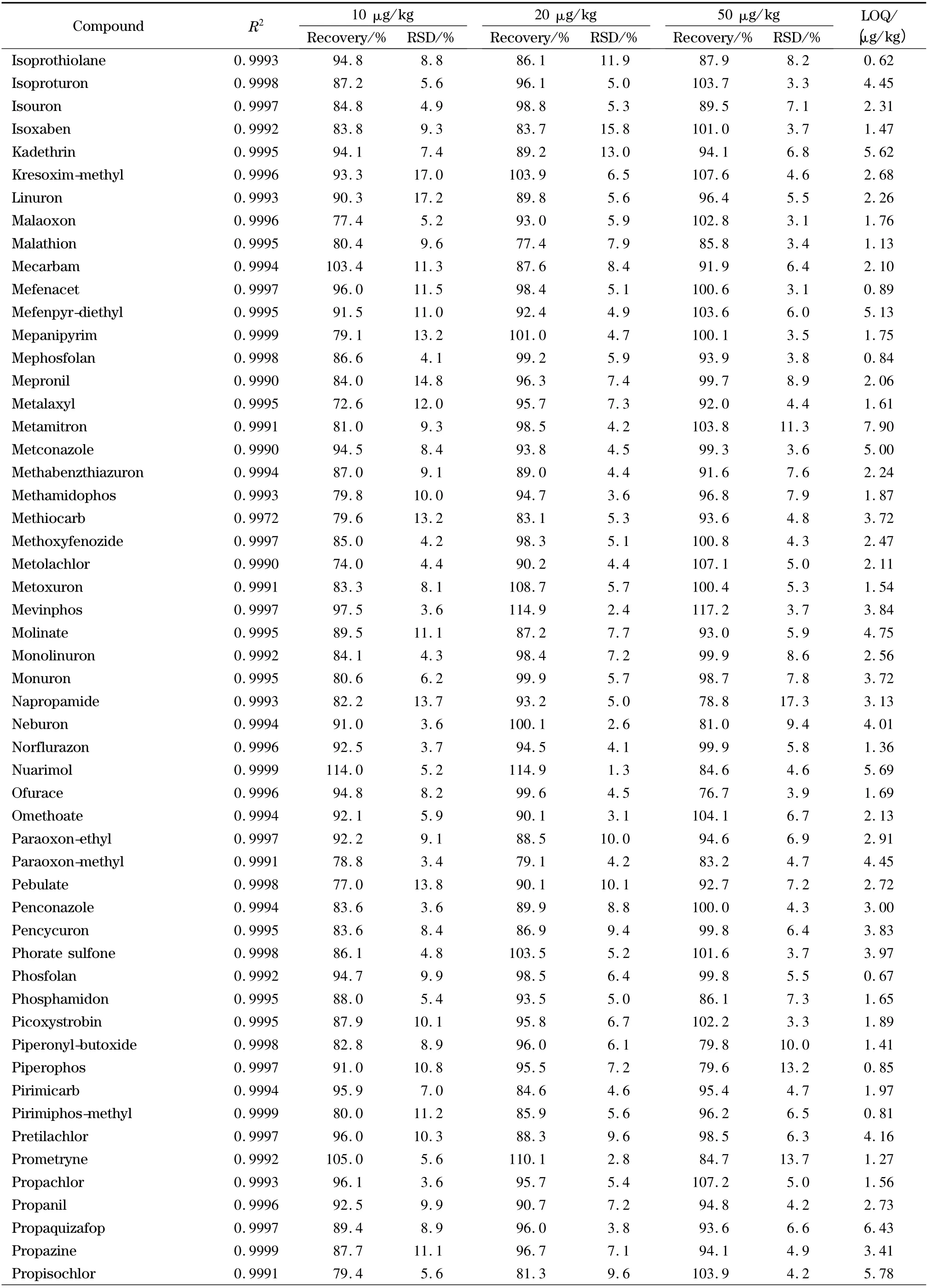
表2 (续)Table 2 (Continued)
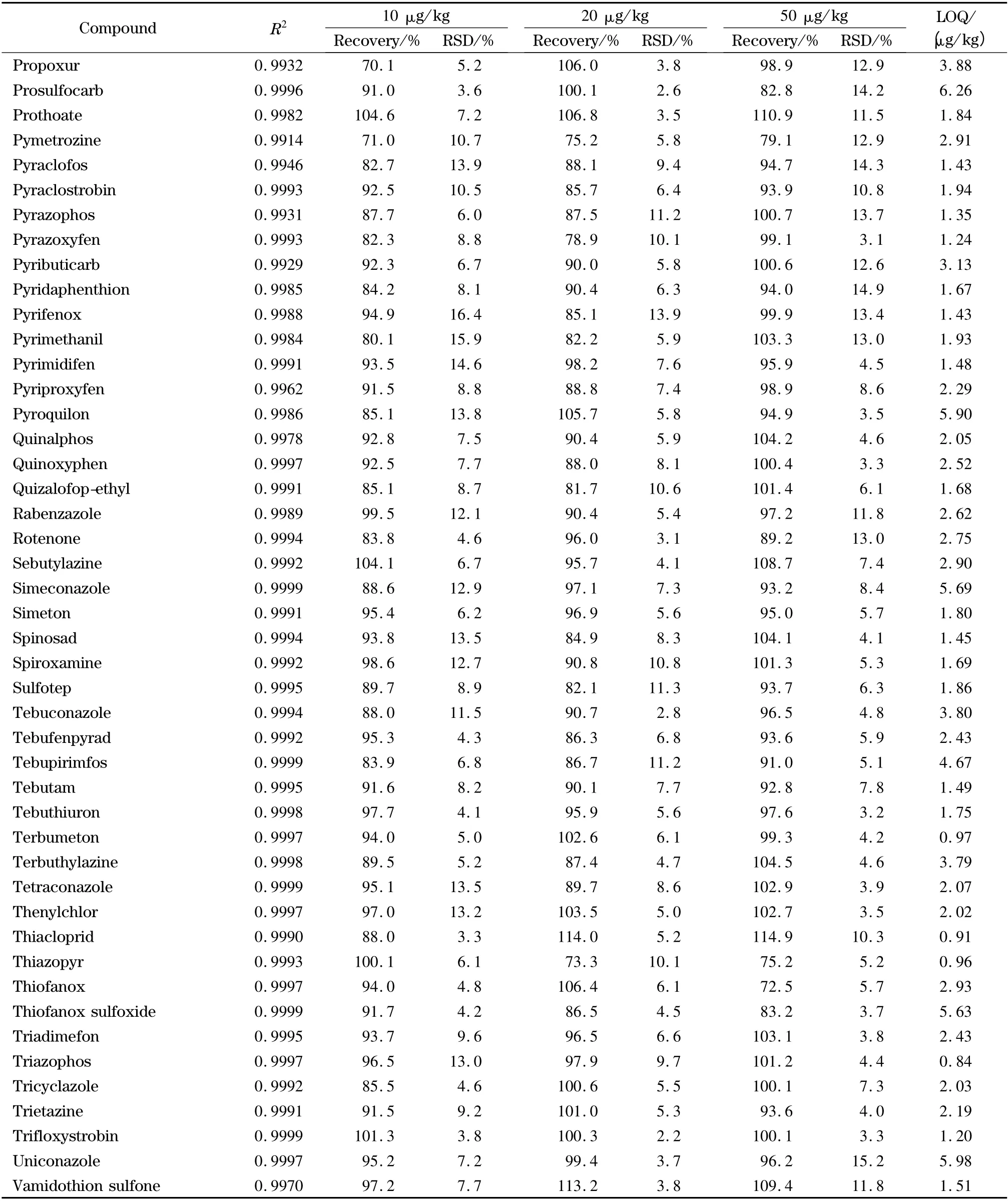
表2 (续)Table 2 (Continued)
欧盟、日本、国际法典委员会(CAC)规定茶叶中农药的MRLs 分别为0.01 ~30.0、0.01 ~30.0、0.10 ~50.0 mg/kg,没有明确规定的农药残留限量均采用0.01 mg/kg 的一律标准[10]。按10 倍信噪比计算定量限,茶叶中204 种农药的定量限均小于10 μg/kg(见表2),满足各国农药残留限量标准的要求。以嘧菌环胺(cyprodinil)、苯线磷亚砜(fenamiphos sulfoxide)、噻唑磷(fosthiazate)、稻瘟灵(isoprothiolane)4 种农药为例,其在接近定量限的加标茶叶样品(添加水平为1 μg/kg)中的提取离子流图见图6。向茶叶空白样品中添加204 种农药的混合标准溶液,添加水平分别为10、20、50 μg/kg,然后按照本实验方法进行提取、净化和检测,每个加标水平重复6 次测定,204 种农药的平均回收率为68.1% ~117.2%,相对标准偏差(RSD)为3.1% ~18.9% (见表2)。
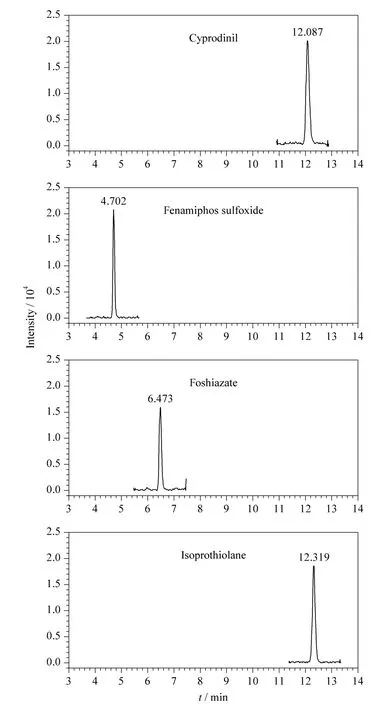
图6 茶叶空白样品中添加水平为1 μg/kg 的嘧菌环胺、苯线磷亚砜、噻唑磷和稻瘟灵的提取离子流图Fig.6 Extracted ion chromatograms of cyprodinil,fenamiphos sulfoxide,fosthiazate and isoprothiolane spiked in blank tea at 1 μg/kg
2.6 实际样品的检测
应用本文所建立的筛查方法对日常送检的10份茶叶样品进行了204 种农药残留的快速筛查检测,检测结果见表3。结果表明,其中的8 份茶叶样品共检出15 种农药。8 份样品中检出农药的一级检索得分均高于70,二级检索得分高于60 的农药占89.7%。低于60 的农药,经背景扣除后,二级得分均提高到60 以上,故达到了确证要求。例如,样品1 中避蚊胺的二级检索得分为49.11,其扣除背景前的镜像对比结果见图7a,背景中主要的特征离子均匹配较好,但由于样品中含有m/z 110.071 3、138.065 9 和195.087 2 共3 个干扰离子,使匹配程度降低,二级检索得分较低。背景扣除后(见图7b),3 个干扰离子被成功扣除,二级检索得分提高为87.22,从而确定含有避蚊胺。为了验证结果,取4 份阳性样品,用GB/T 23205-2008 方法进行再次检测,测得结果与本方法结果基本一致,证明了本方法的有效性。
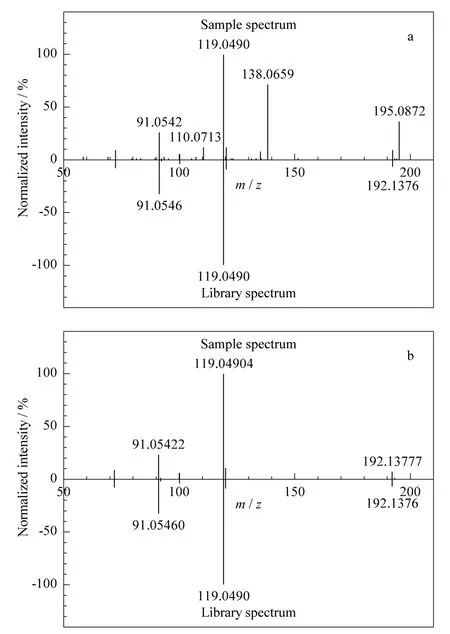
图7 茶叶中避蚊胺(a)扣除背景前和(b)扣除背景后碎片离子的镜像对比结果Fig.7 Comparison of spectral difference results of (a)before and (b)after background subtraction of diethyltoluamide in a tea sample
从表3 可以看出,噻嗪酮、啶虫脒和三唑磷的检出率较高,8 份样品按GB 2763-2014《食品中农药最大残留限量》衡量,所有检出农药均未超标;按欧盟限量标准衡量,只有样品6 中三唑磷的检出含量为54.07 μk/kg,超出了欧盟规定的最大残留限量,其余农药均未超标。由此可知,虽然茶叶样品中农药的检出率较高,但大部分农药的残留水平偏低,满足我国限量要求。
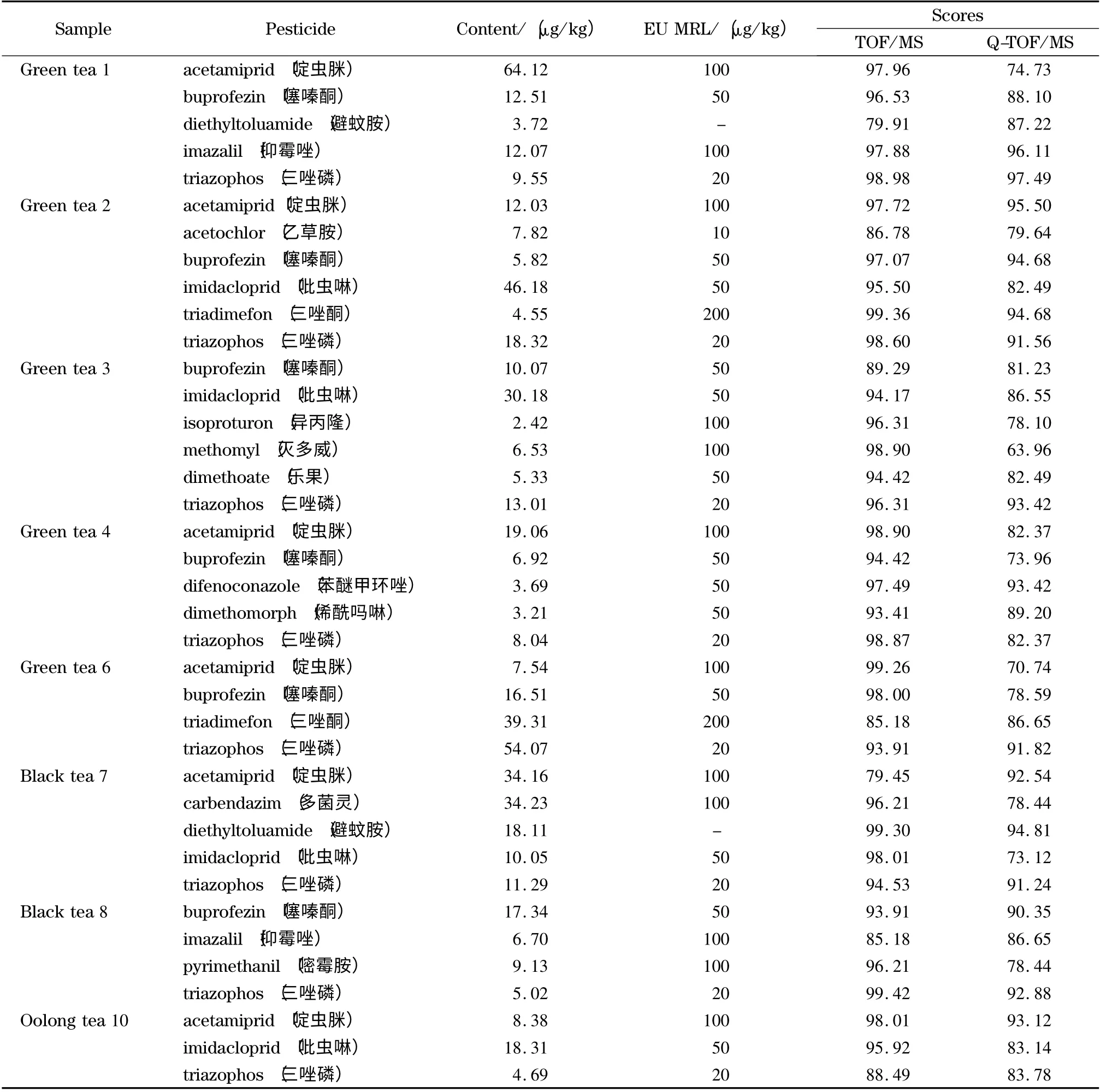
表3 10 份实际样品的测试结果Table 3 Detected results of ten real samples
3 结论
本实验建立了茶叶中204 种农药的UPLC-QTOF/MS 快速筛查方法。利用目标化合物特征离子的精确质量数、保留时间、同位素比例等信息,结合数据库的检索,实现了无需对照标准品同时筛查和确认茶叶中204 种农药。204 种农药在5 ~100 μg/kg 范围线性良好,平均回收率为68.1% ~117.2%。该方法快速、准确,分析通量高,可以为茶叶中多农残的快速筛查和质量控制提供重要的方法依据。
[1] Ma H M,Wang Y Q,Qian H. China Tea Processing (马惠民,王永强,钱和. 中国茶叶加工),2012(4):18
[2] Li Y. [MS Dissertation]. Qinhuangdao:Yanshan University(李岩. [硕士学位论文]. 秦皇岛:燕山大学),2013
[3] GB 2763-2014
[4] Dong J B,Wang J H. Food Science (董金斌,王金花. 食品科学),2009,30(12):230
[5] Deng X J,Qian G,Chen X P,et al. Food Chem,2014,145:853
[6] Mezcua M,Martinez-Uroz A,Fernandez-Alba A. Gas Chromatography. London:Elsevier,2012
[7] Liu B,Zhao H X. Chinese Journal of Analysis Laboratory(刘冰,赵红霞. 分析试验室),2013,32(10):77
[8] Zhao H X,Zhao S C,Deng L G,et al. Food Anal Methods,2013,6:497
[9] Jia W,Huang J R,Ling Y,et al. Journal of Instrumental Analysis (贾玮,黄峻榕,凌云,等. 分析测试学报),2013,32(1):9
[10] Pang G F,Fan C L,Li Y,et al. Journal of Instrumental Analysis (庞国芳,范春林,李岩,等. 分析测试学报),2012,31(9):1017
[11] GB/T 23205-2008
[12] Li X Y,Zhang H Y,Fan C L,et al. Chemistry (李晓颖,张红医,范春林,等. 化学通报),2014,77(2):123
[13] Peng X,Zhao Z Y,Kang J,et al. Chinese Journal of Analysis Laboratory (彭兴,赵志远,康健,等. 分析试验室),2014,33(3):282
[14] Li X J,Peng T,Li C J. Journal of Instrumental Analysis(李晓娟,彭涛,李重九. 分析测试学报),2012,31(5):628
[15] Zhao Z Y,Shi Z H,Kang J,et al. Chinese Journal of Chromatography (赵志远,石志红,康健,等. 色谱),2013,31(4):327
[16] Mezcua M,Malato O,Garcia-Reyes J F,et al. Anal Chem,2009,81(3):913
[17] Zhang F,Yu C T,Wang W W,et al. Anal Chim Acta,2012,757:39
[18] Lou Z Y,Chen Z M,Luo F J,et al. Chinese Journal of Chromatography (楼正云,陈宗懋,罗逢健,等. 色谱),2008,26(5):568
[19] JAP-178 GC/MS
[20] Hou R Y,Bian H Z,Zhao X X,et al. Journal of Instrumental Analysis (侯如燕,卞红正,赵秀霞,等. 分析测试学报),2011,30(1):58
[21] Lou Z Y,Tang F B,Chen Z M,et al. Chinese Journal of Analysis Laboratory (楼正云,汤富彬,陈宗懋,等. 分析试验室),2009,28(Suppl):76
[22] Xu J,Chen J,Ye H Y,et al. Journal of Instrumental Analysis (徐娟,陈捷,叶弘毅,等. 分析测试学报),2011,30(9):990
[23] Feng Y W,Xue Q H,Wang Z J. Food and Fermentation Industries (冯永巍,薛庆海,汪振炯. 食品与发酵工业),2013,39(5):176
[24] 2002/657/EC
[25] Xiang P,Shen M,Zhuo X Y. Journal of Instrumental Analysis (向平,沈敏,卓先义. 分析测试学报),2009,28(6):753
[26] Duan J Y. [MS Dissertation]. Beijing:Chinese Academy of Agricultural Sciences (段俊彦. [硕士学位论文]. 北京:中国农业科学院),2009
猜你喜欢
煤化工(2022年3期)2022-07-08
食品安全导刊(2021年20期)2021-08-30
世界最新医学信息文摘(2021年12期)2021-06-09
色谱(2021年7期)2021-06-07
中华养生保健(2020年10期)2021-01-18
中华养生保健(2020年5期)2020-11-16
当代化工研究(2016年5期)2016-03-20
中国资源综合利用(2016年10期)2016-01-22
实用中西医结合临床(2015年7期)2015-02-28
特产研究(2014年4期)2014-04-10
