
Identi fi cation,synthesis and characterization of process related des fl uoro impurity of ezetimibe and HPLC method validations☆
2015-12-21 00:50:03EsenBellurAticiBekirKarl
Esen Bellur Atici,Bekir Karlığa
Deva Holding A.Ş.,Çerkezköy-2 Production Plant,KaraağaçMh.Fatih Blv.No:26,Address No:2278035833,Kapaklı,Tekirdağ,Turkey
Original Article
Identi fi cation,synthesis and characterization of process related des fl uoro impurity of ezetimibe and HPLC method validations☆
Esen Bellur Atici*,Bekir Karlığa
Deva Holding A.Ş.,Çerkezköy-2 Production Plant,KaraağaçMh.Fatih Blv.No:26,Address No:2278035833,Kapaklı,Tekirdağ,Turkey
A R T I c L E I N F o
Article history:
13 April2015
Accepted 14 April2015
Available online 24 April 2015
Ezetimibe Des fl uoro ezetimibe Synthesis Characterization NMR HPLC
Ezetimibe,which selectively inhibits cholesterol absorption across the intestinal wall and is used as an antihyperlipidemic agent,is synthesized for commercial use as a drug substance in highly pure form. During the synthetic process development studies of ezetimibe,an impurity was detected in the fi nal product at levels ranging from 0.05%to 0.15%in reverse phase gradient high performance liquid chromatography(HPLC)method and its molecular weight was determined by LC–MS analysis.The impurity was identi fi ed as(3R,4S)-3-((S)-3-(4-fl uorophenyl)-3-hydroxypropyl)-4-(4-hydroxyphenyl)-1-phenylazetidin-2-one which is called des fl uoro ezetimibe(lactam-related)impurity,synthesized and characterized,the mechanism of its formation was discussed in detail.After all standardization procedures,it was used as a reference standard during validation of HPLC method and routine analyses.In addition, content of Eze-1 des fl uoro impurity in Eze-1 intermediates was speci fi ed as 0.10%to keep the formation of des fl uoro ezetimibe impurity under control and the related substances HPLC method was validated accordingly.
©2015 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
1.Introduction
There are two recognized sources of cholesterol in the serum: biosynthesis in the liver and absorption of dietary cholesterol in the small intestine[1,2].Statins inhibit 3-hydroxy-3-methylglutaryl coenzyme A(HMG-CoA)reductase,the catalyst of the ratelimiting step of cholesterol biosynthesis in the liver,and have been prescribed as the predominant class of cholesterol-lowering agents since 1980s[3].Ezetimibe,(3R,4S)-1-(4-fl uorophenyl)-3-((S)-3-(4-fl uorophenyl)-3-hydroxypropyl)-4-(4-hydroxyphenyl)
azetidin-2-one(Scheme 1),has become an increasingly important choice for reducing serum cholesterol level and it is the only marketed example of this new class of anticholesterolemia drugs [4,5].Ezetimibe is a potent,metabolically stable cholesterol absorption inhibitor[6],which strongly blocks the absorption of biliary and dietary cholesterol from the small intestine without affecting the absorption of fat-soluble vitamins,triglycerides or bile acids[7].It may be used alone or together with statins(e.g. ezetimibe/simvastatin)for the treatment of primary hypercholesterolemia,homozygous sitosterolemia,homozygous familial hypercholesterolemia,and mixed hyperlipidemia[8,9],and reduces the risk of coronary heart disease[10].
Organic and pharmaceutical chemists were attracted by the novel structure and potent biological activity of ezetimibe,which led to the development of several syntheses for this active pharmaceutical ingredient(API)[11–17].Few analytical methods have been reported in the literature for the determination of ezetimibe including studies on its pharmaceutical dosage forms and degradation studies[18–21].In recent years,studies related to identi fi cation,synthesis and characterization of process and degradation related impurities,including stereoisomers of ezetimibe, have been reported[22–28].
During high performance liquid chromatograph(HPLC)analyses of the drug substance ezetimibe,synthesized according to the process given in Scheme 1[16],one process related impurity was observed consistently in the range of 0.05–0.15%.As per the general guidelines recommended to qualify the drug substance by International Conference on Harmonisation(ICH)[29,30],the acceptable level for a known and unknown related compounds (impurity)should be less than 0.15%and 0.10%,respectively;and the impurities present in the drug substance must be identi fi ed and characterized.In recent years,the impurity pro fi le of a drug substance becomes more important for marketing approval and this work is done as part of a drug development process[31–35].
The impurity observed on HPLC analyses of ezetimibe was identi fi ed by LC–MS studies as des fl uoro ezetimibe,(3R,4S)-3-((S)-3-(4-fl uorophenyl)-3-hydroxypropyl)-4-(4-hydroxyphenyl)-1-phenylazetidin-2-one and pathway of its formation was clari fi ed after thorough evaluation of the ezetimibe synthetic route.Thesame pathway was used for synthesis of des fl uoro impurity and its related intermediates.All compounds synthesized in the scope of this study were characterized by MS,IR and NMR(1H,13C,DEPT,19F).To the best of our knowledge,des fl uoro ezetimibe impurities were mentioned only in a few reports[36–39]and there is no report in the literature on the identi fi cation,synthesis and characterization of the des fl uoro impurity.As complementary of this work,HPLC related substances methods for ezetimibe(Eze)and its intermediate Eze-1 were validated and reported herein.

Scheme 1.Synthesis of ezetimibe and des fl uoro ezetimibe impurity.
2.Experimental
2.1.Chemicals,reagents and samples
Ezetimibe samples were taken from commercial batches produced by Deva Holding A.Ş.(Tekirdag˘,Turkey),or synthesized in the R&D laboratories of Deva.Ezetimibe standard was supplied by a specialized team on standardization of reference standards for analytical use in Deva.Synthetic and analytical reagents and solvents were supplied from different chemical companies such as Merck KGaA(Darmstadt,Germany),J.T.Baker(Phillipsburg,USA), Sigma-Aldrich(St.Louis,MO,USA),Lab-Scan(Gliwice,Poland), Acros Organics(Geel,Belgium),Akkim(Yalova,Turkey),Sodas Sodyum Sanayi A.S.(Izmir,Turkey),and Emekcioglu Tuz A.S. (Ankara,Turkey).All non-aqueous reactions were performed in dry glassware under an atmosphere of dry nitrogen.Deionized water was prepared using Milli Q plus puri fi cation system(Millipore,Bedford,MA,USA).Deuterated solvents(dimethylsulfoxided6 and deuterated chloroform)were purchased from Merck KGaA (Darmstadt,Germany).
2.2.Mass spectrometry
The mass spectra were recorded on Waters LC–MS ZQ 2000/ 4000 system(Waters Corporation,Milford,MA,USA).The fragmentation pro fi le of the samples was established by carrying out MS studies in the positive or negative electrospray ionization(ESI) mode.For LC–MS identi fi cation of the impurity,the HPLC method given infra for ezetimibe related substances was used.The samples of synthesized compounds were directly infused using a syringe at a concentration of 1 mg/mL in acetonitrile.
2.3.NMR spectroscopy
1H,13C and DEPT NMR experiments were performed on a 300 MHz NMR Spectrometer(Varian NMR Instruments,Oxford, UK;later acquired by Agilent Technologies,Santa Clara,CA,USA) using deuterated dimethylsulfoxide(DMSO-d6)and deuterated chloroform(CDCl3)as a solvent and tetramethylsilane(TMS)as an internal standard.
2.4.FT-IR spectroscopy and melting range
Samples were measured as neat by Attenuated Total Refl ectance(ATR)on Shimadzu FTIR Spectrometer IR Prestige-21 (Shimadzu Corporation,Kyoto,Japan)in the range of 600–4000 cm-1with 20 scans and 2 cm-1resolution.Melting ranges of the samples were determined on an Electrothermal 9100 digital melting point instrument(Thermo Fisher Scienti fi c,Essex,UK).
2.5.HPLC
Chromatographic separations were performed on HPLC system with Waters Alliance 2695 separation module equipped with a Waters 996 photodiode array detector and an Empower-pro data handling system(Waters Corporation,Milford,MA,USA).
The related substances analysis of ezetimibe was carried out on Zorbax Rx C8(0.25 m×4.6 mm,5μm)column at 5°C sample and 35°C column temperatures.The injection volume and detection wavelength were fi xed at 10μL and 220 nm,respectively.Data was acquired during 50 min by using gradient elution system fl owing at a rate of 1.3 mL/min.Buffer solution was prepared by dissolving 2.71 g of potassium dihydrogen phosphate in 1000 mL of water and pH was adjusted to 3.0±0.05 with 10%phosporic acid.Mixture of buffer solution and acetonitrile(80:20)was used as mobile phase A;and acetonitrile as diluent and mobile phase B.Test solution was prepared by dissolving 25.0 mg of sample in 25.0 mL of acetonitrile(1.0 mg/mL).Reference solution was prepared by dissolving 25.0 mg of ezetimibe reference standard(RS)in 25.0 mL of acetonitrile,followed by dilution of 1.0 mL of this solution to 100.0 mL with acetonitrile,then dilution of 1.0 mL of the resulting solution to 10.0 mL with acetonitrile(0.10%with respect to the test solution,0.001 mg/mL).Test solutions were injected freshly and reference solution was stable up to 48 h at room temperature.The separation was employed using gradient elution based on the following program:time(min)/%ofmobile phase B:0/12,5/12,25/ 62,40/62,41/12,50/12.This analysis method was also used during standardization of des fl uoro ezetimibe.
The related substances analysis of Eze-1 was carried out on Kromasil(or ACE)C18(0.25 m×4.6 mm,5μm)column with 10μL injection volume at a wavelength of 225 nm by using gradient elution fl owing at a rate of 1.1 mL/min during 25 min.Column and sample temperatures were 30°C and 25°C,respectively.Buffer solution was prepared by dissolving 2.72 g of potassium dihydrogen phosphate in 1000 mL of water and pH was adjusted to 7.5±0.05 with 10%potassium hydroxide.Mixture of buffer solution and acetonitrile(60:40)was used as mobile phase A;and acetonitrile as diluent and mobile phase B.Test solution was prepared by dissolving 20.0 mg of the sample in 50.0 mL of acetonitrile(0.4 mg/mL).Reference solution was prepared by dissolving 20.0 mg of Eze-1 RS in 50.0 mL of acetonitrile,followed by dilution of 1.5 mL of this solution to 50.0 mL with acetonitrile,then dilution of 1.0 mL of the resulting solution to 10.0 mL with acetonitrile(0.30%with respect to the test solution;0.0012 mg/mL). Reference and test solutions were found stable up to 48 h at room temperature.The separation was employed using gradient elution based on the following program:time(min)/%of mobile phase B: 0/0,2/0,15/35,18/35,20/0,25/0.This analysis method was also used during standardization of des fl uoro Eze-1.
The related substances analysis of Eze-3 was carried out on Zorbax Rx C8(0.25 m×4.6 mm,5μm)column with 20μL injection volume at a wavelength of 220 nm by using gradient elution fl owing at a rate of 1.3 mL/min during 40 min.Column and sample temperatures were 35°C and 20°C,respectively.Mobile phase A was prepared by dissolving 2.74 g of potassium dihydrogen phosphate in 1000 mL of water and pH was adjusted to 3.0±0.05 with 10%phosphoric acid.Acetonitrile was used as diluent and mobile phase B.Test solution was prepared by dissolving 10.0 mg of the sample in 10.0 mL of acetonitrile(1.0 mg/mL)and reference solution by dissolving 10.0 mg of Eze-3 RS in 50.0 mL of acetonitrile.Reference and test solutions were found stable up to 96 h at room temperature.The separation was employed using gradient elution based on the following program:time(min)/%of mobile phase B:0/30,5/30,30/70,32/30,40/30.
The related substances analysis of Eze-4 was carried out on Zorbax Rx C8(0.25 m×4.6 mm,5μm)column with 10μL injection volume at a wavelength of 220 nm by using gradient elution fl owing at a rate of 1.3 mL/min during 55 min.Column and sample temperatures were 35°C and 25°C,respectively.Mobile phase A was prepared by dissolving 2.74 g of potassium dihydrogen phosphate in 1000 mL of water and pH was adjusted to 7.5±0.05 with 10%potassium hydroxide.Acetonitrile was used as diluent and mobile phase B.Test solution was prepared by dissolving 30.0 mg of the sample in 10.0 mL of acetonitrile(3.0 mg/mL)and reference solution by dissolving 10.0 mg of Eze-4 RS in 50.0 mL of acetonitrile (0.2 mg/mL).Reference and test solutions were found stable up to 96 h at room temperature.The separation was employed using gradient elution based on the following program:time(min)/%of mobile phase B:0/35,2/35,28/75,46/75,47/35,55/35.
2.6.Ultra performance liquid chromatography(UPLC)
Chromatographic separations for Eze-5,6,7 and their related substances were performed on Waters Acquity UPLC system equipped with Waters TUV detector and Empower-pro data handling system(Waters Corporation,Milford,MA,USA).The analysis was carried out on ChromPEARL SH C18(0.15 m×2.0 mm, 1.7μm)column with 2μL injection volume at a wavelength of 220 nm by using gradient elution fl owing at a rate of 0.4 mL/min during 33 min.Column and sample temperatures were 40°C and 5°C,respectively.Buffer solution was prepared by dissolving 1.36 g of potassium dihydrogen phosphate in 1000 mL of water and pH was adjusted to 7.5±0.05 with 10%potassium hydroxide. Mixture of buffer solution and acetonitrile(80:20)was used as mobile phase A,and acetonitrile as diluent and mobile phase B. Test solution was prepared by dissolving 25.0 mg of the sample in 25.0 mL of acetonitrile(1.0 mg/mL).Reference solution was prepared by dissolving 25.0 mg of related reference standard in 25.0 mL of acetonitrile,followed by dilution of 0.5 mL of this solution to 50.0 mL with acetonitrile(1.0%with respect to the test solution;0.01 mg/mL).Reference and test solutions were found stable up to 48,24 and 72 h at 5°C,respectively.The separation was employed using gradient elution based on the following program:time(min)/%of mobile phase B:0/38,12/53,15/63,25/ 88,30/38,33/38.Analyses of des fl uoro Eze-5,6,and 7 were carried out by using this method.
2.7.Synthesis of ezetimibe and des fl uoro ezetimibe impurity
2.7.1.Synthesis of Eze-1 and des fl uoro Eze-1
4-Hydroxybenzaldehyde(1.0 equiv.)was dissolved in isopropanol at 60°C under stirring.4-Fluoroaniline or aniline (1.0 equiv.)was added to the resulting solution and the mixture was stirred at 60°C for 3 h.The solution was allowed to cool to ambient,during which the expected imine crystallized.The crystals were fi ltered out,washed with isopropanol and dried at room temperature to obtain 4-((4-fl uorophenylimino)methyl)phenol (Eze-1)or 4-((phenylimino)methyl)phenol(des fl uoro Eze-1)as yellowish crystalline powder with 82%and 85%yield,respectively.
2.7.2.Synthesis of Eze-4

Fig.1.HPLC chromatogram of(A)ezetimibe spiked with des fl uoro ezetimibe impurity(system suitability solution)and(B)Eze-1 spiked with des fl uoro Eze-1 impurity and other related impurities(system suitability solution).
Mixture of 3-[5-(4-fl uorophenyl)-1,5-dioxopentyl]-4-(S)-phenyloxazolidin-2-one(Eze-3)(1.0 equiv.),methanol,trimethylorthoformate (2.0 equiv.)and catalytic amount of p-toluene sulfonic acid monohydrate(pTSA)was heated and stirred until no trace of starting material(Eze-3)was observed in the reaction mixture.After completion of the reaction,the mixture was concentrated to a de fi ned volume,quenched by addition of N,N-diisopropylethylamine(DIPEA)and followed by cooling for crystallization.The product crystals were fi ltered,washed with methanol and dried under vacuum to obtain Eze-4 as white to slightly yellowish crystalline powder (83%).
2.7.3.Synthesis of Eze-5 and des fl uoro Eze-5
Mixture of DIPEA(4.0 equiv.),benzyl chloroformate(CbzCl, 1.3 equiv.)and toluene was added into a cooled suspension of Eze-1 or des fl uoro Eze-1(1.2 equiv.)in methylene chloride.In a separate reaction vessel,titanium complex was prepared by mixing titanium tetrachloride(0.9 equiv.)and titanium tetraisopropoxide (0.3 equiv.)in methylene chloride under cooling.Solution of Eze-4 (1.0 equiv.)in methylene chloride was added into the cooled Eze-2 (Cbz-protected Eze-1)or des fl uoro-Eze-2(Cbz-protected des fl uoro Eze-1)solution at-15°C and it was followed by additional cooling to-45°C.The titanium complex mixture was added into this solution at-45°C and stirred for reaction.The reaction was quenched by addition of acetic acid and aqueous solution of citric acid,respectively.The synthesis was followed by warming up the reaction mixture to 25°C,work-up and crystallization processes to obtain Eze-5 or des fl uoro Eze-5 as white to slightly yellowish crystalline powder with 49%and 35%yield,respectively.
2.7.4.Synthesis of Eze-6 and des fl uoro Eze-6
Dimethylsul fi de borane(1 M in methylene chloride,BH3.Me2S) and(R)-2-Me-CBS oxazaborolidine(1 Min toluene)Corey–Bakshi–Shibata catalyst mixture was prepared in methylene chloride.Solution of Eze-5 or des fl uoro Eze-5(1.0 equiv.)in methylene chloride was added into the catalyst mixture.After completion ofthe reaction,it was quenched by addition of methanol and aqueous solution of hydrochloric acid(1.0 M),respectively.The synthesis was followed by work-up and crystallization processes to obtain Eze-6 or des fl uoro Eze-6 as white to slightly yellowish crystalline powder with 79%and 82%yield,respectively.

Fig.2.Structures of ezetimibe,des fl uoro impurity and intermediates.
2.7.5.Synthesis of Eze-7 and des fl uoro Eze-7
Lithium perchlorate(0.1 equiv.)and hexamethyldisilazane (HMDS,0.9 equiv.)were added into the solution of Eze-6 or des fl uoro Eze-6(1.0 equiv.)in methylene chloride at room temperature.After stirring for a de fi ned time period and completion of the reaction,it was quenched by addition of water.The synthesis was followed by work-up and crystallization processes to obtain Eze-7 or des fl uoro Eze-7 as white to slightly yellowish crystalline powder with 90%and 94%yield,respectively.
2.7.6.Synthesis of ezetimibe and des fl uoro ezetimibe
Solution of Eze-7 or des fl uoro Eze-7(1.0 equiv.)intetrahydrofuran(THF)was cooled to-20°C.N,O-bis(trimethylsilyl)acetamide(BSA,2.1 equiv.)and catalytic amount of tetrabutylammonium hydroxide(TBAH,50%in methanol)were added into this mixture and stirred for a de fi ned time.After completion of the reaction,it was quenched by addition of acetic acid and concentrated by distillation.Methanol,acetic acid and catalytic amount of Pd/C catalyst(5%)were added into the residue and stirred in the presence of hydrogen(H2)gas.After completion of the reaction,the mixture was fi ltered,concentrated to a de fi ned volume and crystallized to obtain crude ezetimibe.The synthesis was followed by recrystallization and drying processes to obtain ezetimibe or des fl uoro ezetimibe in highly pure form as white to off-white crystalline powder with 60%and 48%yield,respectively.

Table 1 Purity,melting range,FTIR and mass spectraldata.
3.Results and discussion
3.1.Ezetimibe and des fl uoro ezetimibe syntheses
Ezetimibe is aβ-lactam type hypolipidemic agent bearing three chiralcenters.Of these,two are located on theβ-lactam ring while the last one(alcohol)is present in the side chain.The molecule is manufactured as a single enantiomer with absolute stereochemistry(2R,11S,5S).The enantioselective determination of the opposite OH-enantiomer,which is likely to be detected in the fi nal API,is an integral part of the ezetimibe speci fi cation.A validated chiral HPLC method was used to control this enantiomer with the acceptance limit of not more than 0.15%.
The stereochemistry issues are important for ensuring the production of diastereo as well as enantiopure fi nal API.Ezetimibe and des fl uoro ezetimibe were synthesized according to the reaction steps given in Scheme 1.Ketone functionality of Eze-3 was protected by using trimethylorthoformate and catalytic amount of p-toluenesulfonic acid to form dimethyl ketal Eze-4. Corresponding Eze-1 imines(Schiff base)were prepared by reaction of 4-hydroxybenzaldehyde and 4-fl uoroaniline or aniline and then protected by reaction with benzyl chloroformate under basic conditions to obtain Eze-2 or des fl uoro Eze-2 intermediates,respectively.Highly exothermic coupling reaction of protected imines(Eze-2 or des fl uoro Eze-2)and Eze-4 was done under strong cooling by using titanium complex(Ti(OiPr)Cl3)prepared by mixing titanium tetrachloride and titanium tetraisopropoxide with the ratio of 3:1.The reaction was quenched by acid addition, and ketal deprotection was done by further acid treatment in situ to obtain crystalline Eze-5 or des fl uoro Eze-5.
Chiral centers C-2 and C-11 were created by diastereoselective addition of optically active titanium enolate of Eze-4 to C=N double bond of Cbz-protected imine Eze-2.Chirality of oxazolidide-ketone Eze-4 induces chirality of both C-2 and C-11 chiral centers during the coupling reaction of Eze-2 and Eze-4 intermediates.Thus,it is highly important to have this intermediate with high optical purity.For this reason,optical purity of Eze-3 was controlled and a limit for content of its enantiomer was set as not more than 0.10%and reference standard of Eze-3 enantiomer was prepared.Oxazolidide-acetal Eze-4 was treated with a strong base DIPEA to generate an enolate which by action of Ti (OiPr)Cl3formed Ti(IV)-enolate of Eze-4.The latter exists due to steric bulkiness of phenyl group on oxazolidinone ring exclusively in(Z)-con fi guration.Imine Eze-2 is(E)–con fi gured and binds to Tienolate,thus forming essentially sterically less hindered and sterically more hindered complexes.In both cases,imine is located below the plane of enolate,which de fi nes absolute con fi guration at C-2.Accordingly,only two diastereoisomeric products were formed in this reaction step with highly prevailing as expected(6–8:1).Both primary products were hydrolyzed in situ by acids forming ketone Eze-5 and its diastereoisomer.Luckily,minor and unwanted diastereoisomer of Eze-5 is much more soluble and single crystallization was suf fi cient to reduce its content.

Fig.3.UPLC chromatograms of(A)Eze-5,(B)Eze-6,and(C)Eze-7 spiked with their related des fl uoro impurities.
The chiral center bearing hydroxyl group(C-5)was created by applying Corey–Bakshi–Shibata(CBS)protocol to Eze-5 intermediates to obtain the corresponding Eze-6 alcohols,namely reduction of ketone with borane dimethyl sul fi de complex in the presence of(R)-2-Me-CBS oxazaborolidine((R)-hexahydro-1-methyl-3,3-diphenylpyrrolo[1,2]-c[1,3,2]oxazaborole)as a chirality inductor.The enantioselective reduction of Eze-5 provided highly predominating alcohol Eze-6 accompanied by minor amounts of epimeric alcohol epi-Eze-6 with the amount of maximum 4%.The content of the latter compound was set to not more than 3.0%andreference standard was prepared by CBS-reduction of Eze-5 in the presence of(S)-2-Me-CBS oxazaborolidine.By using solubility differences of epimers,content of minor epimeric alcohol was further decreased by recrystallization of Eze-6 in methanol.Of course,diastereomeric impurity of ketone Eze-5 also underwent reduction to corresponding alcohols.However,the content of such alcohols,which was already low,was further decreased by crystallization of the reaction product Eze-6.

Table 21H and13C NMR assignments for Eze-1 and des fl uoro Eze-1.
In the following step,TMS protection of alcoholmoiety of Eze-6 intermediates was done by reaction with HMDS under catalysis of lithium perchlorate to form corresponding Eze-7 intermediates and puri fi ed by crystallization which enhanced the stereochemical purity further.In the last complex step,trans-β-lactam ring was closed fi rst by the action of BSA and catalytic amount of TBAH upon Eze-7 intermediates.The formation of cis-β-lactam that would originate from silyl-ether originating from epimeric Eze-5 was much more sluggish.This implies that the formation of measurable quantities of ezetimibe stereoisomer with 2R,11R,5S con fi guration as an impurity in fi nal APIis highly improbable.The formed 2-azetidinone intermediates reacted without isolation further for Cbz-and TMS-deprotections by Pd/C catalyzed hydrogenation under acidic conditions to obtain crude ezetimibe or des fl uoro ezetimibe.Furthermore,products were crystallized so that the content of such an impurity with 2R,11R,5S con fi guration, if any formed,was further reduced.2,11-epimer and enantiomer of ezetimibe with 2S,11R,5S and 2S,11R,5R con fi gurations,respectively,would be only hypothetical products of CBS-reduction of very improbable diastereomeric form of ketone Eze-5.5-Epimer of ezetimibe with 2R,11S,5R con fi guration and called ezetimibe OH-isomer was more likely to be detected in manufactured API as it was formed from epimer of Eze-6 via epimer of Eze-7.Accordingly, this stereoisomer was prepared and routinely tested for its presence in fi nal API(limit not more than 0.15%).Ezetimibe and des fl uoro ezetimibe were fi nally recrystallized to reach pharmaceutically acceptable purity and highly pure product to be used as reference standard during validation of related substances HPLCmethod of ezetimibe,respectively.All the facts discussed above clearly demonstrate that the production of stereochemically highly pure fi nal products was ensured.
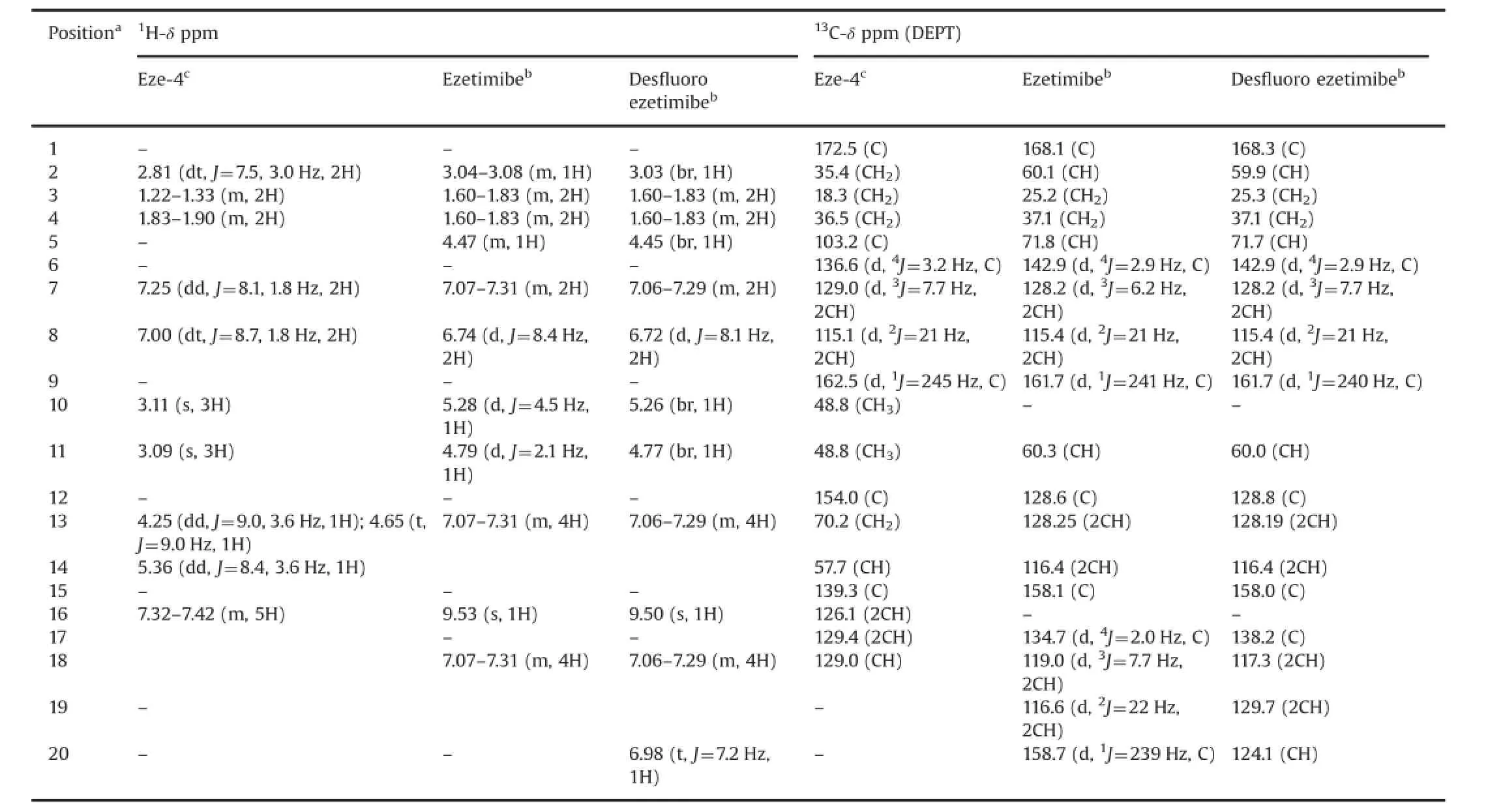
Table 31H and13C NMR assignments for Eze-4,ezetimibe and des fl uoro ezetimibe.

Table 41H NMR assignments for intermediates of ezetimibe and des fl uoro ezetimibe impurity.
3.2.Detection and identi fi cation of des fl uoro impurity
A typical analytical HPLC chromatogram of a production batch of ezetimibe bulk drug spiked with des fl uoro impurity(system suitability solution)is shown in Fig.1A.The impurity was detected in the fi nal drug substance during process development studies and its identi fi cation was done by LC–MS.The ESImass spectrum of the impurity observed at 18.05 min(RRT 0.97)exhibited a molecular ion at m/z 392 in positive ion mode,indicating molecular weight of 391,which was less by 18 amu than that of ezetimibe.The difference relates to a missing fl uorine(-19)and addition of a hydrogen(+1)instead.There are two possibilities to form such an impurity in the fi nal product.One is potential Eze-3 des fl uoro impurity and the second one is Eze-1 des fl uoro impurity. After our analytical studies,it was found that Eze-1 contained the related des fl uoro impurity and Eze-3 did not.So,the related desfl uoro impurity was subsequently synthesized to obtain suf fi cient quantity for full characterization and further analytical studies. The synthesized impurity was co-injected with ezetimibe to confi rm its identity based on the retention time matching.The impurity at 18.05 min(RRT 0.97)was well resolved from ezetimibe and the HPLC method was validated accordingly.
3.3.Formation of des fl uoro impurity and structure elucidations
Solubility ofdes fl uoro ezetimibe impurity is very similar to that of ezetimibe and it was not possible to remove this impurity by recrystallization processes.That is why its formation should be kept under control from the beginning of the ezetimibe production process.The source of this impurity is des fl uoro Eze-1,which is found in the intermediate Eze-1.As shown in Scheme 1,Eze-1 and des fl uoro Eze-1 and their corresponding intermediates are similarly reacting under the same synthetic conditions.So,to keep the amount of des fl uoro impurity within the speci fi ed known impurity limits(0.15%)in the fi nal product,the amount of desfl uoro Eze-1 should be kept below 0.15%in Eze-1.To be on the safe side,the limit of des fl uoro Eze-1 was speci fi ed as 0.10%in Eze-1; an HPLC method for related substances was developed and validated accordingly.Chromatogram of system suitability solution where Eze-1 was spiked with its related substances is given in Fig.1B.
Synthesis of ezetimibe was done by using 0.08%des fl uoro Eze-1 containing Eze-1 intermediate,resulting in formation of 0.05%of des fl uoro ezetimibe impurity.Purity,melting range,IR and mass data of the synthesized compounds(Fig.2)are listed in Table 1. Also,UPLC chromatograms of Eze-5,Eze-6 and Eze-7 spiked with their related des fl uoro impurities are shown in Fig.3.
1H and13C NMR assignments for the structures given in Fig.2 are shown in Tables 2–5.Compared with ezetimibe and its intermediates,1H NMR spectra of des fl uoro compounds showed one more proton peak in the aromatic region.Also,a very dramatic difference could be seen on13C NMR shifts where fl uorine splitting was clearly seen on eight different aromatic carbons and in the case of des fl uoro compounds only four of them were observed and others did not split due to a regular C–H bonding.19F NMR spectrum of ezetimibe showed two multiplets at-116.40 and-118.75 ppm corresponding to the fl uorine atoms on C-9 and C-20(Fig.4),respectively.As expected,des fl uoro ezetimibe showed one multiplet at-116.43 ppm corresponding to the fl uorine atom on C-9.All spectral data(Supplementary Appendix A)con fi rmed the structures of des fl uoro impurities synthesized in scope of this study.

Table 513C NMR chemical shifts for intermediates of ezetimibe and des fl uoro ezetimibe impurity.
3.4.Validation of HPLC methods
Methods were validated according to ICH Q2(R1)guideline [40].
3.4.1.Validation of Eze-1 related substances method
Related substances limits for Eze-1 are speci fi ed as follows: 4-hydroxy benzaldehyde not more than 0.50%,4-fl uoroaniline not more than 0.50%,des fl uoro Eze-1 not more than 0.10%,any other impurity not more than 0.30%and total impurities not more than 1.0%and validated accordingly.Purities of reference standards used during the validation study are as follows:Eze-1 99.7%, 4-hydroxybenzaldehyde 99.8%,4-fl uoroaniline 99.8%,and desfl uoro Eze-1 99.7%.
The system suitability was conducted throughout the validation study by using 1.2μg/mL of Eze-1 reference solution and evaluated by making six replicate injections.The system was deemed to be suitable for use as the relative standard deviation (RSD)of the areas was found below 5.0%,resolution above 2.0(4.6) and symmetry factor below 1.5(1.1).Average of the areas was found as 44,117 with standard deviation(SD)459 and RSD was calculated as 1.0%,con fi rming the injection repeatability of the developed method.
The following injections were done for speci fi city test:two diluents,two from each impurity at speci fi cation limit,two from mixture of impurities at speci fi cation limit,two test solutions (0.4 mg/mL Eze-1),and two test solutions spiked with impurities at speci fi cation limit.No interference was observed in the chromatogram of diluent where Eze-1 and its impurities peaks elute. Purity angles of Eze-1 and all impurities peaks(0.25,0.46,1.04, and 1.76,respectively)were found less than purity thresholds (0.85,0.65,1.23,and 1.97,respectively).Thus,the method was found speci fi c for determination of the related substances in Eze-1.
Stability of solutions was evaluated by injection of two reference solutions and two test solutions freshly and then each 24 h during two days.Reference and test solutions were accepted as stable for a time period during which the absolute differences between fresh and different period results were below 0.10%and 0.05%,respectively.This difference did not exceed 0.01%and 0.04%, respectively for both solutions with RSD value of 0.7%and they were found stable up to 48 h.At longer time periods,amounts of 4-hydroxybenzaldehyde and 4-fl uoroaniline impurities were increased due to the dissociation of Eze-1 to its starting materials.

Fig.4.Comparison of1H,13C and19F NMRs of ezetimibe and des fl uoro ezetimibe impurity.
Limit of detection(LOD)and limit of quantitation(LOQ)values were calculated according to the following equations:LOD=3× (BN/H)×C;LOQ=10×(BN/H)×C,where BN is baseline-noise (mV),H is height(mV)and C is concentration of the reference solution(μg/mL).Six replicate diluent injections were done for baseline noise calculations,and solution containing Eze-1 RS and impurities at 1.2μg/mL concentrations was injected six times for calculation of peak heights.LOD and LOQ values for Eze-1,4-hydroxybenzaldehyde,4-fl uoroaniline and des fl uoro Eze-1 were found to be 0.003%(0.013μg/mL),0.003%(0.010μg/mL),0.004% (0.014μg/mL),0.003%(0.011μg/mL),and 0.011%(0.042μg/mL), 0.008%(0.032μg/mL),0.012%(0.046μg/mL),0.010%(0.038μg/mL), respectively.LOQ values were below the reported level 0.05%with RSD of 0.3%,0.3%,0.2%and 0.2%,respectively.Signalto noise ratios at LOD and LOQ concentrations were found below 3 and 10,respectively,and met the validation criterion.Precision at LOQ level was done by six replicate injections of Eze-1 RS and impurities solution prepared at LOQ concentration.Averages of the areas were found as 1357,1225,1399 and 1343 with standard deviation of 61,60,94,and 83,and RSD of 4.5%,4.9%,6.7%,and 6.2%,respectively,which were below the acceptance limit of 10.0%.Correction response factors(CRF)were calculated according to the following equation:CRF=(A×Ci)/(Ai×C),where A is peak area of Eze-1,Ai is peak area of the impurity,C is concentration of Eze-1 (μg/mL),and Ci is concentration of the impurity(μg/mL).CRFs for 4-hydroxybenzaldehyde,4-fl uoroaniline and des fl uoro Eze-1 were found as 1.1,1.3,and 0.9,respectively.Chromatograms of diluent, system suitability solution,reference and test solutions,mixture solution of all impurities and test solution spiked with all impurities at speci fi cation limits,LOD and LOQ solutions are given in Fig.5.
Linearity test solutions of Eze-1 RS and its impurities were prepared at fi ve concentration levels ranging from disregard limit (DL)to 150%of the speci fi cation limit and each sample was injected three times.The peak area versus concentration data was analyzed with least squares linear regression.Correlation coef ficient(r)and y-intercept ratio to the response of target concentration were found as 1.0000,0.9999,0.9993,0.9993(≥0.99) and 0.24%,0.95%,0.45%,0.41%(≤5.0%),respectively,indicating the good linearity of the method.Solutions at level1(DL)and level 5(150%of speci fi cation limit)were injected six times,and RSD values of areas were calculated for range study,and found to be 0.7%,1.3%,1.3%,0.8%for level 1 and 0.5%,0.6%,0.4%,0.8%for level 5,respectively,below the acceptance limits of 7.0%and 5.0%.
The accuracy of the method was evaluated on test solutions spiked with Eze-1 impurities in triplicate at three concentration levels of 50%,100%and 150%of the speci fi cation limit.Good-toexcellent recoveries of the impurities(98.63–104.2%)at each level were achieved within the limit range of 90.0–110.0%(Table 6). RSDs of recoveries were found below 5.0%,as 2.0%,2.6%,and 2.0%, respectively.
The precision of the method was investigated by injecting six individualtest solutions of Eze-1(0.4 mg/mL).The same procedure was applied for the inter-day precision by a different analyst using different batch column and different instrument located within the same laboratory.The system suitability results for the intraday and inter-day precision studies were obtained as RSD 3.0%and 0.2%,resolution 4.6 and 3.7,symmetry factor 1.1 and 1.2,respectively.Results obtained for RSD were below 5.0%in both studies (Table 6)and difference between mean of results of two studies was found below 0.05%(absolute value),and the method was found to be suf fi ciently precise since no signi fi cant variation in the found amounts was observed on any day.
Robustness of the method was tested by changing column,fl ow rate by±10%(1.1±0.1 mL/min),buffer pH by±0.2 units (7.5±0.2),column temperature by±3°C(30±3°C),and acetonitrile content of mobile phase A by±5%(40±5%).System suitability parameters were ful fi lled in all of the above varied chromatographic conditions.Difference between the results of normal and altered conditions did not exceed 0.01%(<0.05%), indicating the robustness of the method.

Fig.5.Chromatograms of(A)diluent,(B)reference solution,(C)system suitability solution,(D)solution ofimpurities at speci fi cation limit,(E)test solution,(F)test solution spiked with impurities at speci fi cation limit,(G)LOD solution,and(H)LOQ solution of Eze-1 related substances HPLC method validation.
Requirements for each stage of the validation study were ful fi lled.Thus,our method was found precise,linear,accurate, sensitive,selective and robust,and could be used for related substances analysis of Eze-1.This method was used in routine productions of ezetimibe to control impurities in the intermediate Eze-1 and keep the drug substance impurity content in acceptable range.
3.4.2.Validation of ezetimibe related substances method
Related substances limits for ezetimibe are speci fi ed as follows: des fl uoro ezetimibe not more than 0.15%,any impurity individually not more than 0.10%and total impurities not more than 0.5%and validated accordingly.Purities of reference standards used during the validation study are as follows:ezetimibe 99.9% and des fl uoro ezetimibe 97.9%.
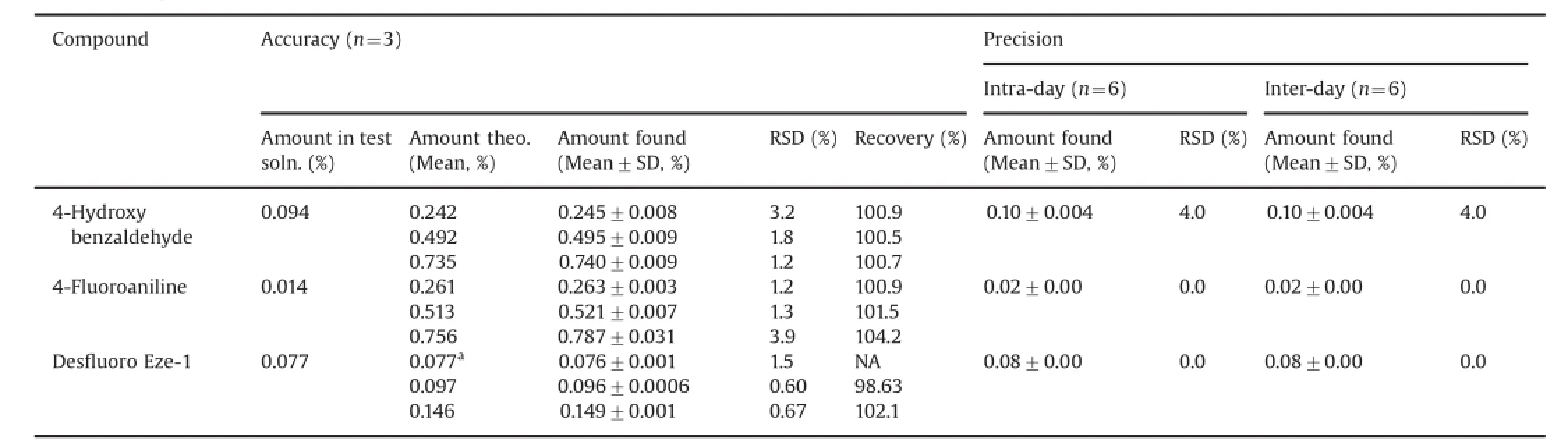
Table 6 Accuracy and precision studies of the HPLC method for determination of Eze-1 related substances.
System suitability was conducted throughout the validation study by using ezetimibe standard solution in concentration of 1.0μg/mL and evaluated by making six replicate injections.The system was accepted to be suitable for use as RSD of areas was below 5.0%,symmetry factor in the range of 0.8–1.5,and the resolution between peaks of ezetimibe and des fl uoro ezetimibe was below 2.0.Average of the areas was found as 14,165 with standard deviation of 411.RSD of areas,symmetry factor and resolution were found as 2.90%,1.0 and 3.5,respectively,con fi rming the system suitability of the developed method.
Speci fi city test was carried out by injections of three diluents, and two from each of the following solutions:test,des fl uoro ezetimibe at speci fi cation limit and test solution spiked with des fl uoro ezetimibe at speci fi cation limit(0.10%).No peak was observed on diluent chromatogram at the retention time of ezetimibe and des fl uoro ezetimibe.Purity angles of ezetimibe and des fl uoro ezetimibe peaks were found less than purity thresholds. Stability of reference solution was evaluated by injection of two different reference solutions freshly and then each 24 h during two days.Stability of test solutions was evaluated by injection of two different sample solutions freshly and then each 3 h during 24 h.Solutions were accepted as stable for a time period during which the difference between fresh and different period results was below 0.05%.The reference and test solutions were found stable up to 48 h and 24 h,respectively.
The LOD and LOQ for ezetimibe and des fl uoro ezetimibe were estimated at a signal-to-noise ratio of 3:1 and 10:1,respectively,by injecting six replicate diluent and solution containing ezetimibe and des fl uoro ezetimibe at 1μg/mL concentration.The LOD and LOQ for ezetimibe and des fl uoro ezetimibe were found 0.012% (0.12μg/mL),0.013%(0.13μg/mL)and 0.041%(0.41μg/mL),0.043% (0.43μg/mL),respectively.LOD and LOQ signal to noise ratios were found to be 3.4,3.1(≥3)and 10.7,10.4(≥10),respectively.LOQ was found below the reported level 0.05%and met the validation criterion.Precision at the LOQ level was done by injecting six replicates of solution containing ezetimibe and des fl uoro ezetimibe at LOQ concentration.Average of the areas for ezetimibe and des fl uoro ezetimibe was found as 4879 and 5171 with standard deviation of 65 and 52,and RSD value of 1.3%and 1.0%,which were below the acceptance limit of 10.0%.CRF was calculated according to the following equation:CRF=(A×Ci)/(Ai×C),where A is peak area of ezetimibe,Ai is peak area of des fl uoro impurity,C is concentration of ezetimibe(1μg/mL),and Ci is concentration of desfl uoro impurity(1μg/mL).RSD of the peak areas was found to be 1.7%and CRF for des fl uoro impurity was calculated as 1.01.Chromatograms of diluent,system suitability solution,reference and test solutions,des fl uoro impurity and test solution spiked with des fl uoro impurity at speci fi cation limit(0.15%),LOD and LOQ solutions are given in Fig.6.
Linearity test solutions were prepared at fi ve concentration levels ranging from DL to 120%of the speci fi cation limit(0.15%) and each sample was injected three times.The peak area versus concentration data was analyzed with least squares linear regression.Correlation coef fi cient(r)and y-intercept ratio to the response of target concentration for ezetimibe and des fl uoro impurity were found as 0.9930,0.9998(above 0.99)and 0.94%,1.72% (below 5.0%),respectively,indicating the good linearity of the method.Solutions at level 1 and level 5 were injected six times and RSDs of ezetimibe and des fl uoro ezetimibe peak areas were calculated for range study,and found as 1.8%,1.0%and 0.6%,0.4%, respectively,below the acceptance limit of 10.0%and 5.0%.
The accuracy of the method was evaluated by injection of six des fl uoro ezetimibe solutions at speci fi cation limit,six different test solutions for determination of des fl uoro impurity content,and three test solutions spiked with des fl uoro ezetimibe at concentration levels of80%,100%and 120%of the speci fi cation limit in triple.Good-to-excellent recoveries(100.4%–100.8%)at each level were achieved within the limit range of90.0%– 110.0%(Table 7).RSD of recoveries and RSD of des fl uoro ezetimibe peak areas for six replicate injections were found as 1.4%(below 5.0%)for both.
The method precision of an analytical procedure expresses the close agreement between single results of the method applied repeatedly by the same analyst,using the homogeneous sample, the same instrumentation and same reagents.The precision of the method was investigated by injecting six individual test solutions. The inter-day precision was conducted according to the same procedure on a different day by a different analyst using different batch column and different instrument located in the same laboratory.No difference was found between the results of intra-day and inter-day precision studies and the amount of des fl uoro impurity was found to be 0.09%with RSD of 0.0%on both days(Table 7),indicating the precision of the method.
The robustness of an analytical method is a measure of the effect of variable conditions on analytical results in the process of analysis.For the determination of the method’s robustness,fl ow rate,pH of the buffer and column temperature were varied within realistic range and the effect of variations on analytical procedure was examined and reported.Robustness of the method was tested by changing fl ow rate(1.3±0.13 mL/min),buffer pH(3.0±0.3), and column temperature(35±3.5°C)by±10%.In allof the above varied chromatographic conditions,RSD and symmetry factors were found in the range of1.5%–2.8%and 0.8– 1.0,respectively.No difference was found between the results of normal and altered conditions,suggesting the robustness of the method.

Fig.6.Chromatograms of(A)diluent,(B)reference solution,(C)system suitability solution,(D)solution of des fl uoro ezetimibe impurity at speci fi cation limit,(E)test solution,(F)test solution spiked with des fl uoro ezetimibe impurity at speci fi cation limit(0.15%),(G)LOD solution(ezetimibe 0.12μg/mL(0.012%)and des fl uoro impurity 0.13μg/mL(0.013%)),and(H)LOQ solution(ezetimibe 0.41μg/mL(0.041%)and des fl uoro impurity 0.43μg/mL(0.043%))of ezetimibe related substances HPLC method validation.
System suitability parameters were ful fi lled in each stage of the validation study and the results showed that our developed method is speci fi c,linear,precise,accurate and robust,and it could be used for related substances analyses of ezetimibe manufacturing batches.This method was used in routine productions and stability testing of ezetimibe drug substance.The mount of desfl uoro impurity in ezetimibe was found below the speci fi ed limit and as expected it was not affected during the stability studies since it was not a degradation product.
4.Conclusion
In conclusion,a process related impurity of ezetimibe,produced according to the synthetic route given in Scheme 1,was identi fi ed,synthesized and characterized.Structural elucidations of all synthesized compounds were done by using NMR(1H,13C, DEPT,19F),IR and MS spectral data.Thus,the regulatory requirement was ful fi lled by characterizing this impurity and the prepared impurity standard was used during analytical methodvalidation studies.This work also supported the optimization stage of the process development and enabled us to see and control the critical points of the process.The knowledge of the impurity source(Eze-1)helped us to control the amount of desfl uoro Eze-1 impurity by a newly developed and validated HPLC method,which allowed us to reduce the des fl uoro ezetimibe impurity successfully even below 0.10%in the fi nal drug substance.

Table 7 Accuracy and precision studies of the HPLC method for determination of ezetimibe related substances.
Acknowledgments
The authors are thankful for the management of Deva Holding A.S.,Istanbul,Turkey,and also former owner of the company Zentiva–part of Sano fi-aventis,Tekirdag˘,Turkey for supporting this work and the Scienti fi c and Technological Research Council of Turkey(TUBITAK-TEYDEB Project No:3080507)for the fi nancial support.
Appendix A.Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.jpha.2015.04.002.
References
[1]D.A.Burnett,β-Lactam cholesterol absorption inhibitors,Curr.Med.Chem.11 (2004)1873–1887.
[2]S.D.Turley,J.M.Dietschy,Sterolabsorption by the smallintestine,Curr.Opin. Lipidol.14(2003)233–240.
[3]J.Earl,P.Kirkpatrick,Ezetimibe,Nat.Rev.Drug Discovery 2(2003)97–98.
[4]C.Gagne,H.E.Bays,S.R.Weiss,et al.,Ef fi cacy and safety of ezetimibe added to ongoing statin therapy for treatment of patients with primary hypercholesterolemia,Am.J.Cardiol.90(2002)1084–1091.
[5]J.W.Clader,Ezetimibe and other azetidinone cholesterol absorption inhibitors, Curr.Top.Med.Chem.5(2005)243–256.
[6]S.B.Rosenblum,T.Huynh,A.Afonso,et al.,Discovery of 1-(4-fl uorophenyl)-(3R)-[3-(4-fl uorophenyl)-(3S)-hydroxypropyl]-(4S)-(4-hydroxyphenyl)-2-azetidinone(SCH 58235):a designed,potent,orally active inhibitor of cholesterol absorption,J.Med.Chem.41(1998)973–980.
[7]M.van Heek,H.Davis,Pharmacology of ezetimibe,Eur.Heart J.Suppl.4 (Suppl.J)(2002)J5–J8.
[8]J.Patel,V.Sheehan,C.Gurk-Turner,Ezetimibe(Zetia):a new type of lipidlowering agent,BUMC Proc.16(2003)354–358.
[9]J.W.Clader,The discovery of ezetimibe:a view from outside the receptor,J. Med.Chem.47(2004)1–9.
[10]A.L.Catapano,Ezetimibe:a selective inhibitor of cholesterol absorption,Eur. Heart J.Suppl.3(Suppl.E)(2001)E6–E10.
[11]R.M.Castaner,L.A.Sorbera,J.Castaner,Drugs Ezetimibe,Future 25(2000) 679–685.
[12]D.A.Burnett,Asymmetric synthesis and absolute stereochemistry of cholesterol absorption inhibitor,SCH 48461,Tetrahedon Lett.35(1994)7339–7342.
[13]G.Wu,Y.Wong,X.Chen,et al.,a novel one-step diastereo-and enantioselective formation of trans-azetidinones and its application to the total synthesis of cholesterol absorption inhibitors,J.Org.Chem.64(1999) 3714–3718.
[14]M.S´nieżek,S.Stecko,I.Pan fi l,et al.,Total synthesis of ezetimibe,a cholesterol absorption inhibitor,J.Org.Chem.78(2013)7048–7057.
[15]M.Michalak,M.Stodulski,S.Stecko,et al.,A formalsynthesis of ezetimibe via cycloaddition/rearrangement cascade reaction,J.Org.Chem.76(2011) 6931–6936.
[16]M.Slavikova,H.Stepankova,J.Zezula,et al.,Method of producing(3R,4S)-1-(4-fl uorophenyl)-3-[(3S)-3-(4-fl uorophenyl)hydroxypropyl]-4-(4-hydroxyphenyl)-2-azetidinone,Zentiva K.S.,Czech Republic,PCT Pat.Appl.WO 2009140932 A2(Nov 26,2009).
[17]M.Adiyaman,E.Bellur Atici,Method for preparation of(3R,4S)-1-(4-fl uorophenyl)-3-[(3S)-3-(4-fl uorophenyl)-3-hydroxypropyl]-4-(4-hydroxyphenyl) azetidin-2-one(ezetimibe)and intermediates thereof,Zentiva Kimyasal Urunler San.ve Tic.A.S.,Turkey,TR Pat.Appl.TR 201000116 A1(Jul 21,2011).
[18]R.Sistla,V.S.S.K.Tata,Y.V.Kashyap,et al.,Development and validation of a reversed-phase HPLC method for the determination of ezetimibe in pharmaceutical dosage forms,J.Pharm.Biomed.Anal.39(2005)517–522.
[19]S.Singh,B.Singh,R.Bahuguna,et al.,Stress degradation studies on ezetimibe and development of a validated stability-indicating HPLC assay,J.Pharm. Biomed.Anal.41(2006)1037–1040.
[20]K.R.Chimalakonda,V.Gudala,M.Gutta,et al.,Development and validation of chiral HPLC method for identi fi cation and quanti fi cation of(R)-enantiomer in ezetimibe,Am.J.Anal.Chem.3(2012)478–483.
[21]M.I.Beludari,K.V.Prakash,G.K.Mohan,RP-HPLC method for simultaneous estimation of rosuvastatin and ezetimibe from their combination tablet dosage form,Int.J.Chem.Anal.Sci.4(2013)205–209.
[22]K.Filip,K.Bankowski,K.Sidoryk,et al.,Physicochemical characterization of ezetimibe and its impurities,J.Mol.Struct.991(2011)162–170.
[23]Y.Ren,R.J.Li,Y.Deng,et al.,First synthesis and characterization of SRR/RSS-ezetimibe,Tetrahedron Lett.54(2013)6443–6446.
[24]B.Raman,B.A.Sharma,R.Butala,et al.,Structural elucidation of a processrelated impurity in ezetimibe by LC/MS/MS and NMR,J.Pharm.Biomed.Anal. 52(2010)73–78.
[25]Z.Santa,J.Koti,K.Szoke,et al.,Structure ofthe major degradant ofezetimibe, J.Pharm.Biomed.Anal.58(2012)125–129.
[26]S.Guntupalli,U.K.Ray,N.Murali,et al.,Identi fi cation,isolation and characterization of process related impurities in ezetimibe,J.Pharm.Biomed.Anal. 88(2014)385–390.
[27]Y.Ren,Y.J.Duan,R.J.Li,et al.,First synthesis and characterization of key stereoisomers related to ezetimibe,Chin.Chem.Lett.25(2014)1157–1160.
[28]K.Chimalakonda,V.Kamani,M.Gutta,et al.,Isolation and characterization of R-enantiomer in ezetimibe,Am.J.Anal.Chem.4(2013)488–495.
[29]Guidance for industry Q3A(R2),Impurities in new drug substances,in:International Conference on Harmonisation,2006.
[30]Guidance for industry Q3B(R2),Impurities in new drug products,in:International Conference on Harmonisation,2006.
[31]D.Zhang,X.Song,J.Su,Isolation,identi fi cation and characterization of novel process-related impurities in fl upirtine maleate,J.Pharm.Biomed.Anal.90 (2014)27–34.
[32]M.Dousa,J.Srbek,S.Rádl,et al.,Identi fi cation,characterization,synthesis and HPLC quanti fi cation of new process-related impurities and degradation products in retigabine,J.Pharm.Biomed.Anal.94(2014)71–76.
[33]A.Darcsi,G.Tóth,J.Kökösi,et al.,Structure elucidation of a process-related impurity of dapoxetine,J.Pharm.Biomed.Anal.96(2014)272–277.
[34]S.Thomas,S.K.Paul,S.C.Joshi,et al.,Identi fi cation,synthesis and characterization of an unknown process related impurity in eslicarbazepine acetate active pharmaceutical ingredient by LC/ESI-IT/MS,1H,13C and1H-1H COSY NMR,J.Pharm.Anal.4(2014)339–344.
[35]E.Bellur Atici,B.Karlıg˘a,Identi fi cation,synthesis and characterization of process related impurities ofbenidipine hydrochloride,stress-testing/stability studies and HPLC/UPLC method validations,J.Pharm.Anal.(2015)10.1016/j. jpha.2015.02.001.
[36]V.B.R.Uppala,P.R.Vaddadi,V.V.Sunkara,et al.,Preparation of ezetimibe,U.S. Pat.Appl.US 20070049748 A1(Mar 01,2007).
[37]A.K.Gajjar,V.D.Shah,Impurity pro fi ling:a case study ofezetimibe,Open Conf. Proc.J.2(2011)108–112.
[38]E.L.Regalado,P.Zhuang,Y.Chen,et al.,Chromatographic resolution of closely related species in pharmaceuticalchemistry:dehalogenation impurities and mixtures of halogen isomers,Anal.Chem.86(2014)805–813.
[39]E.L.Regalado,R.K.Dermenjian,L.A.Joyce,et al.,Detection of dehalogenation impurities in organohalogenated pharmaceuticals by UHPLC-DAD-HRESIMS,J. Pharm.Biomed.Anal.92(2014)1–5.
[40]ICH Q2(R1),Validation of analyticalprocedures,in:International Conference on Harmonisation,2005.
14 October 2014
in revised form
☆Peer review under responsibility of Xi'an Jiaotong University.
.Tel.:+90 282 7581771 4413;fax:+90 282 7581770.
E-mail addresses:ebellur@deva.com.tr,esenbellur@yahoo.com(E.Bellur Atici).
 Journal of Pharmaceutical Analysis2015年6期
Journal of Pharmaceutical Analysis2015年6期
- Journal of Pharmaceutical Analysis的其它文章
- Application of analytical instruments in pharmaceutical analysis
- Comparative study of adsorptive role of carbonaceous materials in removal of UV-active impurities of paclitaxel extracts☆
- In vitro–in vivo studies of the quantitative effect of calcium, multivitamins and milk on single dose cipro fl oxacin bioavailability☆
- Optimization,validation and application of an assay for the activity of HMG-CoA reductase in vitro by LC–MS/MS☆
- Antimicrobial and antiproliferative prospective of kosinostatin–a secondary metabolite isolated from Streptomyces sp.☆
- Quanti fi cation of tolvaptan in rabbit plasma by LC–MS/MS:Application to a pharmacokinetic study☆