
HPLC-MSMS测定小柴胡颗粒中柴胡皂苷a与柴胡皂苷d
2015-12-14 01:16:28王嘉林王斯坦
安徽医药 2015年3期
王嘉林,王斯坦
(1.河南省洛阳市食品药品检验所,河南洛阳 471023;2.无锡瑞年实业有限公司,江苏无锡 214092)
小柴胡颗粒为小柴胡汤的成方现代制剂,小柴胡汤由柴胡、黄芩、半夏、党参、生姜、甘草、大枣组成,来源于东汉名医张仲景《伤寒论》,主要功用为解表散热,疏肝和胃,用于外感病邪犯少阳证。小柴胡颗粒收载于《中国药典》2010年版药典一部,柴胡在方中占30%,为君药,为小柴胡颗粒指标性药材,药典只对柴胡进行定性鉴别,含量测定只是对黄芩苷进行测定,柴胡中主要含有挥发油、黄酮及皂苷类,其中柴胡皂苷a(SSa)与柴胡皂苷d(SSd)为药典中柴胡药材项下质量评价指标,结构式见图1,据报道柴胡皂苷具有抗炎、解热、镇痛、保肝的作用[1-3],SSa与SSd遇酸与光不稳定,易使环氧醚键开环[4],转变为柴胡皂苷 b1、b2。小柴胡颗粒制法遵循古方煎煮,挥发油部分并未含在内,而经过煮制并制粒后柴胡复方中SSa与SSd的含量如何评价成为亟需解决的问题,由于柴胡皂苷无紫外吸收,已有文献对柴胡皂苷测定大多采用203~210 nm波长处进行测定[5-6],末端吸收下色谱峰干扰严重,笔者参照文献[7],采用电喷雾串联质谱对复杂柴胡组方小柴胡颗粒中柴胡皂苷进行含量测定,方法简单可靠,可有效控制小柴胡颗粒的质量。
1 材料
1.1 仪器 Agilent 1260高效液相色谱,G1312B二元高压泵,G1322A真空在线脱气机,G1329B自动进样器,G1316A柱温箱,G6410A三重四极杆串联质谱仪(US),Masshunter工作站,Metter Toledo XP205十万分之一电子分析天平(Swizerland)。
1.2 试药 柴胡皂苷a对照品(中国药品生物制品检定所,批号110777-201309,供含量测定用,含量92.0%),柴胡皂苷d(中国药品生物制品检定所,批号 110778-201208,供含量测定用,含量94.6%),小柴胡颗粒(购自市售不同厂家共6个批次),甲醇(DIKMA 色谱纯),乙腈(MERCK 色谱纯),乙酸铵(FLUKA色谱纯)。
2 方法与结果
2.1 色谱条件 液相条件:Agilent ZORBAX SB C18色谱柱(4.6 mm × 100 mm,3.5 μm),流动相:乙腈—0.05%乙酸铵(40∶60),流速:0.4 mL·min-1,柱温:30℃,进样量3μL。
质谱条件:电喷雾离子源(ESI),毛细管电压3 300 V,干燥器温度 340℃,干燥气流量 12 L·min-1,雾化器压力40 psi,其他质谱参数见表1。

表1 柴胡皂苷a与柴胡皂苷d质谱参数
2.2 样品溶液的制备
2.2.1 对照品储备液的制备 精密称取柴胡皂苷a与柴胡皂苷d约10 mg,置10 mL量瓶中,加70%甲醇溶解,并定容至刻度,作为对照品储备液,放冰箱-20℃下冷冻。
2.2.2 样品溶液的制备 精密称取小柴胡颗粒样品1 g置锥形瓶中,加5%氨水的甲醇溶液50 mL,称定,超声处理30 min,放冷,补足减失重,摇匀,过滤,精密移取2 mL续滤液置25 mL量瓶中,用5%氨水的甲醇溶液定容至刻度,即得供试品溶液。
2.3 方法学考察
2.3.1 阴性样品 依照药典处方比例缺柴胡制备阴性样品,同时依照“2.2.2”项下样品处理方法处理,进样,得阴性样品TIC图,无干扰。见图2。
2.3.2 标准曲线的制备 精密量取1 mL“2.2.1”项下对照品储备液至100 mL量瓶,加70%甲醇定容至刻度,摇匀,作为工作储备液。同步稀释成10、20、50、100、250、500、1 000 μg·L-1的标准曲线溶液。按照2.1项下的色谱及质谱条件进样,以样品浓度X为横坐标,以质谱响应面积Y为纵坐标,拟合曲线得,柴胡皂苷a的线性回归方程为Y=9.785 X-0.510 2,r=0.999 0,柴胡皂苷 d 的线性回归方程为 Y=31.516X-3.16,r=0.991 2。见图 2。
2.3.3 重复性实验 取同一批样品,按“2.2.2”项下制备方法,平行制备6份供试品溶液(样品编号1),分别进样3μL,柴胡皂苷a的RSD为1.84%,柴胡皂苷d的RSD为1.91%。
2.3.4 稳定性试验 取同一份供试品溶液(样品编号1),分别于0、4、8、12、16、24 h 进样,每次进样3μL,柴胡皂苷a的RSD为2.0%,柴胡皂苷 d的RSD为1.58%,表明供试品在24 h内稳定。
2.3.5 精密度实验 精密量取“2.3.2”项下柴胡皂苷混合对照品溶液(10,100,1 000 μg·L-1水平浓度)3μL,按照“2.1”项下的色谱质谱条件分别连续进样6针,计算所得定量离子RSD面积,计算RSD,结果见表2,表明精密度良好。
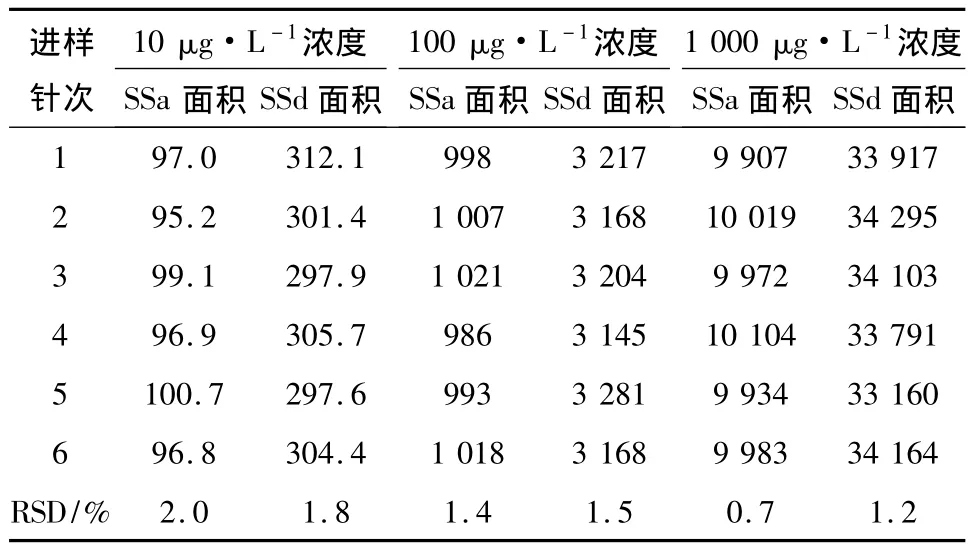
表2 精密度实验表
2.3.6 回收率试验 精密称取已知含量的小柴胡颗粒样品(样品编号2)1 g,共6份,分别精密加入一定量的柴胡皂苷a和d的对照品,按照“2.2.2”项下的方法制备供试品溶液并测定,计算回收率。结果平均回收率柴胡皂苷a(n=6)为99.1%,RSD为1.6%,柴胡皂苷 d(n=6)为 98.7%,RSD 为1.4%。结果见表3。
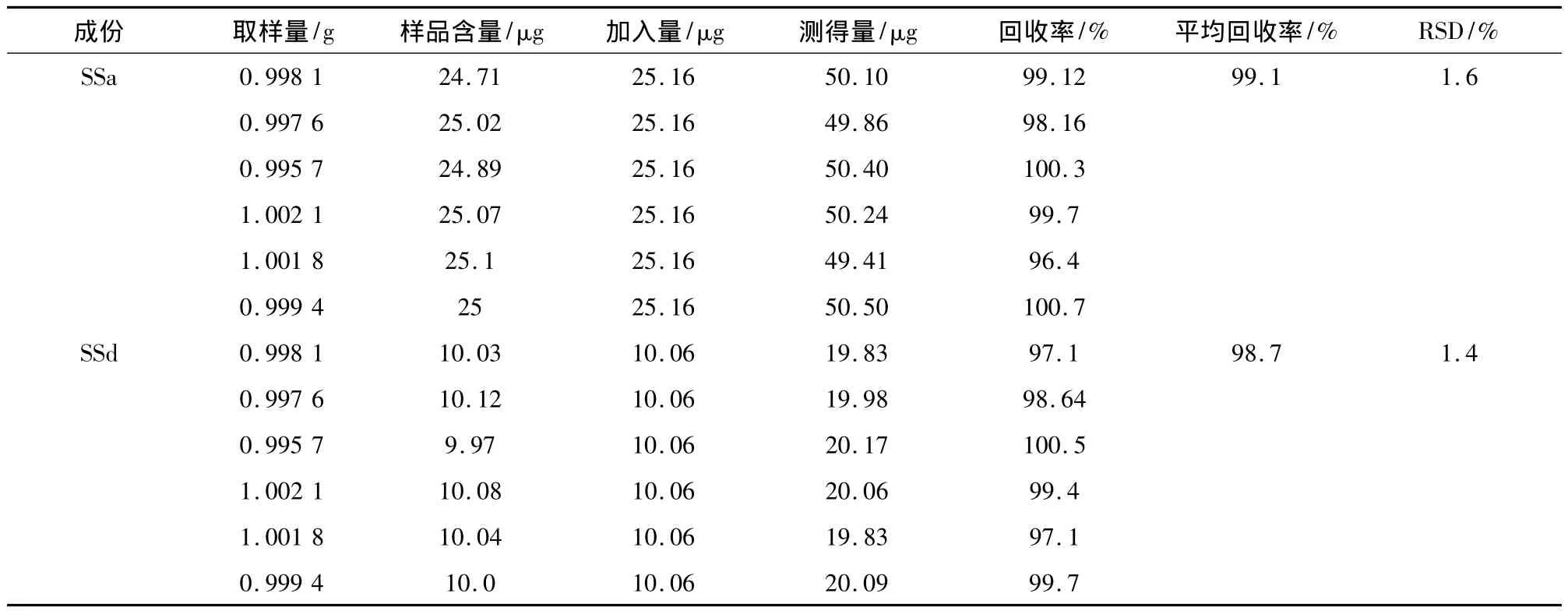
表3 小柴胡颗粒加样回收率结果(n=6)
2.3.7 样品的测定 取所购6个不同批次的小柴胡颗粒样品,按“2.2.2”项下方法制备供试液,按“2.1”项下的色谱质谱条件进样测定,进样3μL,计算含量结果见表4。
3 讨论
3.1 柴胡皂苷的质谱裂解行为 在方法优化时进样柴胡皂苷a与柴胡皂苷d对照品,得到丰度最高的峰为m/z 803峰,分析为[M+Na]+峰,选择负离子模式则加钠峰丰度减小,柴胡皂苷a与d都连接有葡萄糖与半乳糖,增加CID电压,柴胡皂苷a与d发生不同裂解,柴胡皂苷a失去一个葡萄糖得到丰度最高离子m/z 617,与第二丰度m/z 161.1离子,而柴胡皂苷d得到第一丰度离子m/z 617与定性离子m/z 145,两个化学成分定量离子相同,但定性离子不同,故对样品测定不产生干扰。
3.2 提取溶剂的选择 柴胡皂苷a与d在酸性条件下容易向柴胡皂苷b1、b2转化,而小柴胡颗粒制剂中有酸性成分,为避免在处理过程中导致柴胡皂苷发生转化,参照药典提取溶剂采用5%氨水的甲醇溶液作为提取溶剂。

表4 小柴胡颗粒中柴胡皂苷a与d含量测定结果
3.3 提取方式的选择 有报道采用SPE固相萃取进行皂苷类的提取[8],为了除杂增加检测灵敏度,而液质联用采用MRM模式,优势就是在复杂基质条件下对于所测组分进行准确含量测定,故笔者采用超声30 min,即可对柴胡皂苷提取完全
3.4 指标成分的选择 已有文献报道对柴胡成方制剂中柴胡皂苷b1与b2进行含量测定[9-10],但是作者分析柴胡皂苷b1与b2为柴胡皂苷a与d转化而来,转化过程中与制剂工艺及样品稳定性与包装,存放环境等诸多因素相关,而对柴胡皂苷a与d的含量测定可以对小柴胡颗粒的整个制剂工艺以及包装材料选择与存放等进行考察,故依旧选择柴胡皂苷a与d作为其含量测定指标性成分。
3.5 色谱条件的选择 药典柴胡项下对柴胡皂苷a与d的含量测定采用梯度洗脱,低紫外波长进行测定,目的是为了排除紫外干扰,对柴胡皂苷a与d进行完全分离,而笔者采用液质联用,即使柴胡皂苷a与d不能完全分离也不影响其定量,故采用等度洗脱,增加方法的耐用性。
3.6 柴胡皂苷稳定性探讨 据文献研究在酸性条件提取[11],SSa与SSd全部转化。而常温提取时,SSa转化为柴胡皂苷b1,SSd转化为b2;加热时SSa与SSd发生糖苷键酸水解。在弱碱性条件下,SSa与SSd均比较稳定;在中性常温条件提取时,柴胡皂苷a、d比较稳定,但以水作溶剂加热时,SSd转化为柴胡皂苷b2。而有研究表明[12],使提取环境呈弱碱性可以抑制柴胡皂苷环氧醚键的断裂,但是同时需控制碱性的强度,防止乙酰类柴胡皂苷脱去乙酰基,从而达到控制原型皂苷的目的。而在制剂工艺中采用水煎,复方中柴胡及黄芩等药材均含有有机酸,而煎煮温度及有机酸均会造成原生柴胡皂苷向次生皂苷的转化。而利用液质联用的高灵敏度,控制原生皂苷SSa与SSd的含量,进而控制制剂工艺及制剂稳定性对于小柴胡颗粒成方制剂的质量是合理的。
初期质谱条件优化时,在负模式情况下加响应较正模式大,故采用乙酸铵作为流动相添加剂,通过比较 Thermo Hypersil ODS,Merck Star,ZORBAX SB等品牌色谱柱,最终ZORBAX SB色谱柱分离效果最优,故选择ZORBAX SB作为分离色谱柱。
从检测结果来看,市场中小柴胡颗粒的含量差异巨大,最高含量是最低含量的近10倍,分析原因可能与柴胡投料有关,近期由于柴胡价格飞涨,不良厂家可能为了减少原料成本,投料达到柴胡定性要求即可,而柴胡皂苷含量低的另外一个原因与成药放置时间有关,建议厂家在产品上市后对小柴胡颗粒中的有效成分,特别是主要成分稳定性进行考察。建议增加药典中小柴胡颗粒柴胡主要成分柴胡皂苷a与d的质量控制,提高我国传统中药质量。
[1]黄 伟,孙 蓉.柴胡皂苷类成分化学与药理和毒理作用研究进展[J].中药药理与临床,2010,26(3):71-74.
[2]黄幼异,黄 伟,孙 蓉.柴胡皂苷对肝脏的药理毒理作用研究进展[J].中国实验方剂学杂志,2011,17(17):298-301.
[3]马海燕,马玉奎.柴胡皂苷治疗胃溃疡作用的研究[J].齐鲁药事,2011,30(6):318-319.
[4]陈 莹,谭玲玲,蔡 霞.柴胡属植物化学成分研究进展[J].中国野生植物资源,2006,25(2):4-7.
[5]霍务贞,卫世杰.HPLC法测定感冒清口服液中柴胡皂苷a、d的含量[J].中国药房,2013,24(8):741-743.
[6]卫 昊,刘 清,卫伟光.HPLC法测定秦岭柴胡及其不同提取物中柴胡皂苷 a和柴胡皂苷 d[J].中成药,2013,35(2):342-345.
[7]Lee J,Yang DH,Suh JH.Species discrimination of Radix Bupleuri through the simultaneous determination of ten saikosaponins by high performance liquid chromatography with evaporative light scattering detection and electrospray ionization mass spectrometry[J].Jof Chromatogr B Analyt Technol Biomed Life Sci,2011,879(32):3887-3895.
[8]谭 珍,黄婉锋,张 柳.SPE-HPLC法测定小柴胡片中柴胡皂苷 a 的含量[J].广东药学院学报,2007,23(5):511-512.
[9]潘 莉,叶蓓蓓,王伯涛.小柴胡颗粒中柴胡皂苷含量评价方法研究[J].药物分析杂志,2010,30(6):1007-1011.
[10]易润青,宋粉云.小柴胡颗粒中柴胡皂苷a和柴胡皂苷d的毛细管电泳法测定[J].中国医药工业杂志,2012,43(1):47-50.
[11]闵宇航,王京辉,范妙璇.柴胡饮片皂苷类成分变化及质量控制研究[J].药物分析杂志,2014,34(5):836-843.
[12]郭 智,彭 冰,李宗阳.UPLC-QTof-MS测定不同提取条件下柴胡皂苷a和柴胡皂苷d的转化规律[J].天然产物研究与开发,2014,26(5):716-720.
猜你喜欢
遵义医科大学学报(2023年1期)2023-02-06 02:18:40
基层中医药(2022年8期)2022-11-17 08:43:14
家庭医学(下半月)(2020年7期)2020-04-18 13:45:31
中成药(2018年4期)2018-04-26 07:13:09
中成药(2017年8期)2017-11-22 03:18:49
北方牧业(2016年1期)2016-12-17 19:08:50
中国中医药现代远程教育(2014年16期)2014-03-01 04:28:33
中成药(2014年11期)2014-02-28 22:29:44
中国医药生物技术(2014年4期)2014-01-23 09:24:24
天然产物研究与开发(2014年5期)2014-01-09 07:38:50
