
猪肉β-受体激动剂UPLC-MS/MS检测方法建立与应用
2015-12-13 10:39李广兴韩溪吴岩由轩王军连魏菁
东北农业大学学报 2015年12期
李广兴,韩溪,,吴岩,由轩,王军连,魏菁
(1.东北农业大学动物医学学院,哈尔滨 150030;2.黑龙江出入境检验检疫局,哈尔滨 150090;3.黑龙江出入境检验检疫局技术中心,哈尔滨 150090;4.大连出入境检验检疫局,辽宁大连 116001)
猪肉β-受体激动剂UPLC-MS/MS检测方法建立与应用
李广兴1,韩溪1,2,吴岩3,由轩2,王军连2,魏菁4
(1.东北农业大学动物医学学院,哈尔滨 150030;2.黑龙江出入境检验检疫局,哈尔滨 150090;3.黑龙江出入境检验检疫局技术中心,哈尔滨 150090;4.大连出入境检验检疫局,辽宁大连 116001)
建立超高效液相色谱串联质谱法(UPLC-MS/MS)检测猪肉中溴布特罗、西布特罗、西马特罗、克伦特罗等13种β-受体激动剂残留方法。选用盐酸葡萄糖醛苷酶/芳基硫酸酯酶水解,混合型阳离子交换固相萃取净化,以0.1%甲酸水溶液-0.1%甲酸乙腈溶液-甲醇为流动相,超高效液相色谱串联质谱法检测,在0.3、1.0、10 ng·mL-1浓度添加水平,空白肌肉组织中13种药物添加平均回收率范围68.3%~97.2%,日内变异系数范围6.6%~16.9%,日间变异系数范围8.8%~23.3%。该方法检测限为0.1 ng·mL-1,定量限为0.3 ng·mL-1,具有准确敏感特性。在黑龙江省采集202份猪肉样品验证检测13种β-受体激动剂,样品均未检出残留物。
β-受体激动剂;残留检测;超高效液相色谱串联质谱法
网络出版时间2015-12-25 13:11:00[URL]http://www.cnki.net/kcms/detail/23.1391.S.20151225.1311.036.html
李广兴,韩溪,吴岩,等.猪肉β-受体激动剂UPLC-MS/MS检测方法建立与应用[J].东北农业大学学报,2015,46(12):45-51.
Li Guangxing,Han Xi,Wu Yan,et al.Establishment and application of UPLC-MS/MS method detectingβ-agonist drugs in pork meat[J].Journal of Northeast Agricultural University,2015,46(12):45-51.(in Chinese with English abstract)
β-受体激动剂类药物又称“瘦肉精”,食品动物应用起源于美国,20世纪末传至中国,广泛用于养殖业。动物饲料中添加此类药物使动物体内营养物质再分配,显著提高瘦肉比例,然而含有此类药物残留的食品会对人体健康产生极大危害,甚至危及生命[1]。随着人们对其危害性认识,我国已于2002年颁布法规禁止在畜禽养殖中使用β-受体激动剂类药物,但至今这类药物违法添加事件仍屡禁不止,并且出现多种类型β-受体激动剂类药物。我国已将其列为动物食品中重点监控药物,农业部和国家质检总局每年均制定食品安全风险监控计划,监控动物食品中β-受体激动剂类药物残留[2]。测定β-受体激动剂常用方法有免疫分析法[3],利用抗原与抗体的特异性结合反应,具有高选择性、高灵敏度等特点,但易产生假阳性[4];液相色谱法[5-6]、气相色谱-质谱联用法(Gas chromatography mass spectrometry,GC/MS)[7-9]、液相色谱-串联质谱联用法(Liquid chromatographytandem mass spectrometry,LC-MS/MS)[10-11]等,由于β-受体激动剂种类繁多,差异性较大,如采用GC/ MS和HPLC法检测灵敏度低,检测药物种类较少、重现性差,难以满足多种该类药物残留的同时确证检测。本文建立超高效液相色谱串联质谱(Ultra performanceliquidchromatographytandemmass spectrum,UPLC-MS/MS)方法并应用于β-受体激动剂检测,提高分析速度和准确性,降低成本,减少废液产生,为今后相关研究提供理论依据。
1 材料与方法
1.1试剂
1.1.1供试药剂
溴布特罗(Brombuterol,BRB)、西布特罗(Cimbuterol,CIB)、马布特罗(Mabuterol,MAB)、特布他林(Terbutaline,TBL)、西马特罗(Cimatero1,CIMA)、沙丁胺醇(Salbutamo1,SAL)、菲诺特罗(Fenotero1,FEN)、氯丙那林(Clorprenaline,CLP)、莱克多巴胺(Ractopamine,RAC)、克仑特罗(Clenbutero1,CLEN)、妥布特罗(Tulobutero1,TULB)、喷布特罗(Penbutolo1,PEB)和齐帕特罗(Zilpaterol,ZIP)等13种标准品,纯度均大于98.0%,购自德国Dr.Ehrenstorfer公司;盐酸葡萄糖醛苷酶/芳基硫酸酯酶购自美国Merck公司;乙腈、甲酸为色谱纯购自Fisher公司;乙酸铵、乙酸、高氯酸、氢氧化钠、乙酸乙酯、叔丁基甲醚、氨水均为分析纯;所用水为超纯水。
1.1.2供试猪肉样品
动物肌肉样品取自黑龙江省哈尔滨市区、呼兰区、青冈县、双城区、绥化地区等大型肉类屠宰企业。
1.2方法
1.2.1标准溶液制备
分别准确称取13种β-受体激动剂类标准品10 mg于烧杯中,加入少量乙腈,完全溶解后转移至100 mL容量瓶中,用乙腈定容至刻度,配制浓度100 mg·L-1的单标储备液,-20℃保存。分别移取5 mL各单标储备液于100 mL容量瓶中,用初始流动相定容至刻度,配制浓度为5 mg·L-1混合标准储备液,-20℃保存。移取适量混合标准储备液于容量瓶中,用初始流动相定容至刻度,逐级稀释成100、50、25、10和5 μg·L-1混合标准工作液,4℃保存备用。
1.2.2样品处理
准确称取2 g样品于50 mL离心管内,加入0.2 mol·L-1乙酸铵缓冲液(pH 5.2)8.0 mL和盐酸葡萄糖醛苷酶/芳基硫酸酯酶40 μL,涡旋混匀,37℃下避光水浴振荡16 h。酶解后放置至室温,涡旋混匀,10 000 r·min-1高速离心10 min后倾出上清液,置于另一50 mL离心管内,加入0.1 mol·L-1高氯酸溶液5 mL,涡旋混匀,用高氯酸调pH至1.0±0.2,涡旋混匀,10 000 r·min-1离心10 min后,将上清液再转移至另一50 mL离心管内。用10 mol·L-1NaOH溶液调pH至9.5,加入乙酸乙酯15 mL,振荡10 min,5 000 r·min-1离心5 min,将上层有机相转移至另一离心管内。再在下层水相中加入叔丁基甲醚10 mL,振荡10 min,5 000 r·min-1离心5 min。合并有机相,于50℃下氮气吹干,用2%甲酸水溶液5 mL溶解后备用。
将MCX固相萃取柱依次用甲醇、水、2%甲酸水溶液各3 mL活化,取备用液全部过柱,再依次用2%甲酸水溶液、甲醇各3 mL淋洗,抽干,3%氨水甲醇溶液2.5 mL洗脱;将洗脱液于50℃下氮气吹干。残余物用甲醇-0.1%甲酸溶液(体积比为10∶90) 0.2 mL溶解,涡旋混匀,15 000 r·min-1高速离心10 min。取适量上清液,供UPLC-MS/MS测定。
1.2.3色谱质谱条件
Waters ACQUITY UPLCTM BEH C18色谱柱;柱温40℃;样品室温度25℃;进样体积5 μL;流动相A为乙腈,流动相B为0.1%甲酸溶液;流速为0.25 mL·min-1,梯度洗脱程序见表1;运行时间16min。
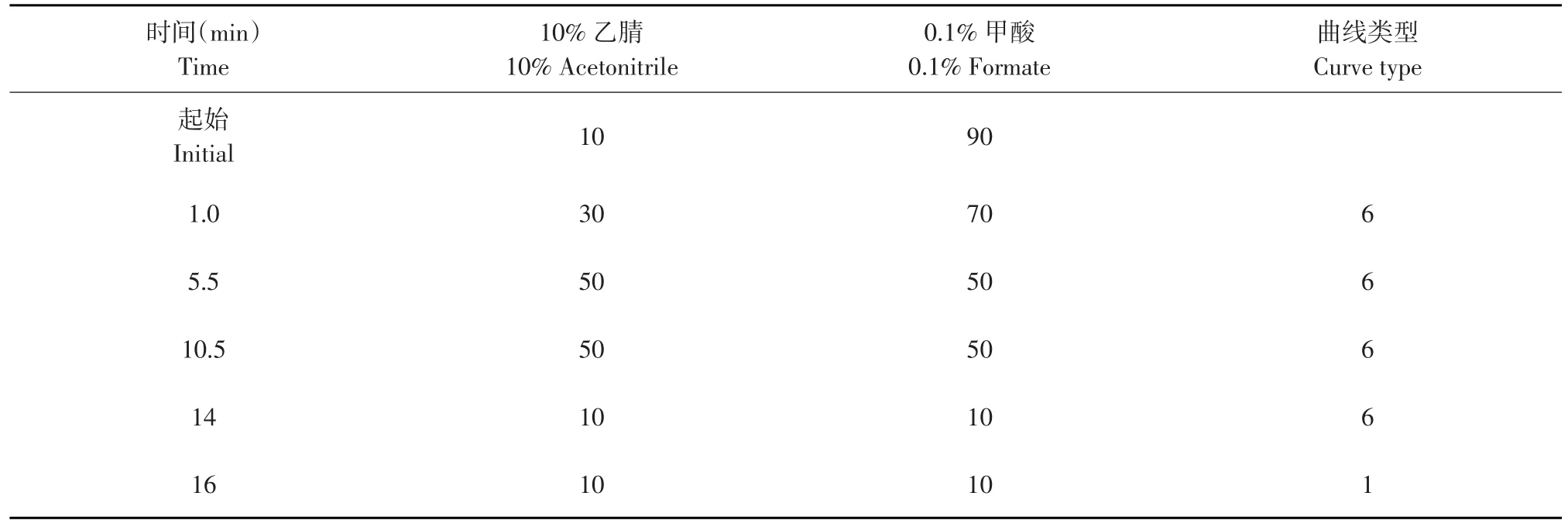
表1 梯度洗脱程序Table 1Gradient elution procedure
电喷雾离子源(ESI)的设置见表2;正离子扫描(ESI+);多反应监测模式(MRM);毛细管电压:3.0 kV;源温度:150℃;脱溶剂温度:400℃;脱溶剂气流量(氮气):650 L·h-1;锥孔气流量(氮气):50 L·h-1;碰撞气流量(氩气):0.20 mL·min-1。光电倍增器电压为650 V。
2 结果与分析
2.1线性范围和标准曲线
准确量取13种β-受体激动剂混合标准工作液,分别添加到2 g空白组织中,制得含量为0.05、0.1、0.2、1.0、50 ng·mL-1系列空白添加试样,按1.2.2所述方法处理,然后上机测定。以各药物定量离子的色谱峰面积为纵坐标(y)、添加浓度(ug·kg-1)为横坐标(x)绘制标准曲线,求出回归方程和相关系数(R),结果表明检测浓度在0.1~50 ng·mL-1范围内呈线性关系,相关系数均在0.99以上(见表3)。
2.2准确度和精密度
称取充分匀浆猪肉空白样品4份,每份5 g(精确到±0.01g),每种样品第一份作空白,其他3份空白组织中添加100 μL对应浓度为0.3、1.0、10 ng·mL-1的13种β受体激动剂标准工作液,即制得高、中、低3个浓度添加样品。按照1.2.2操作,取5 μL进样作回收率试验,每一浓度做6批,每批做6个平行样品分析测定,并计算回收率,试验连续5 d,求出日内和日间变异系数以检验试验精密度。结果表明空白肌肉组织13种药物添加平均回收率范围68.3%~97.2%,日内变异系数范围6.6%~16.9%,日间变异系数范围8.8%~23.3%(见表4)。
2.3灵敏度
灵敏度用最低检测限和最低定量限考查。根据信噪比(S/N)值确定最低检测限和最低定量限。在空白基质中分别添加5个不同水平的13种β-受体激动剂标准溶液,配制不同梯度浓度溶液,测定其信号(以峰高计)与噪声的比值,S/N≥3时的最低浓度为最低检测限,S/N≥6时且回收率和相对标准偏差均符合残留检测方法要求时的最低浓度为最低定量限。此方法检测限均为0.1 ng·mL-1,定量限为0.3 ng·mL-1。空白猪肉添加13种药物10 ng·mL-1色谱图见图1。
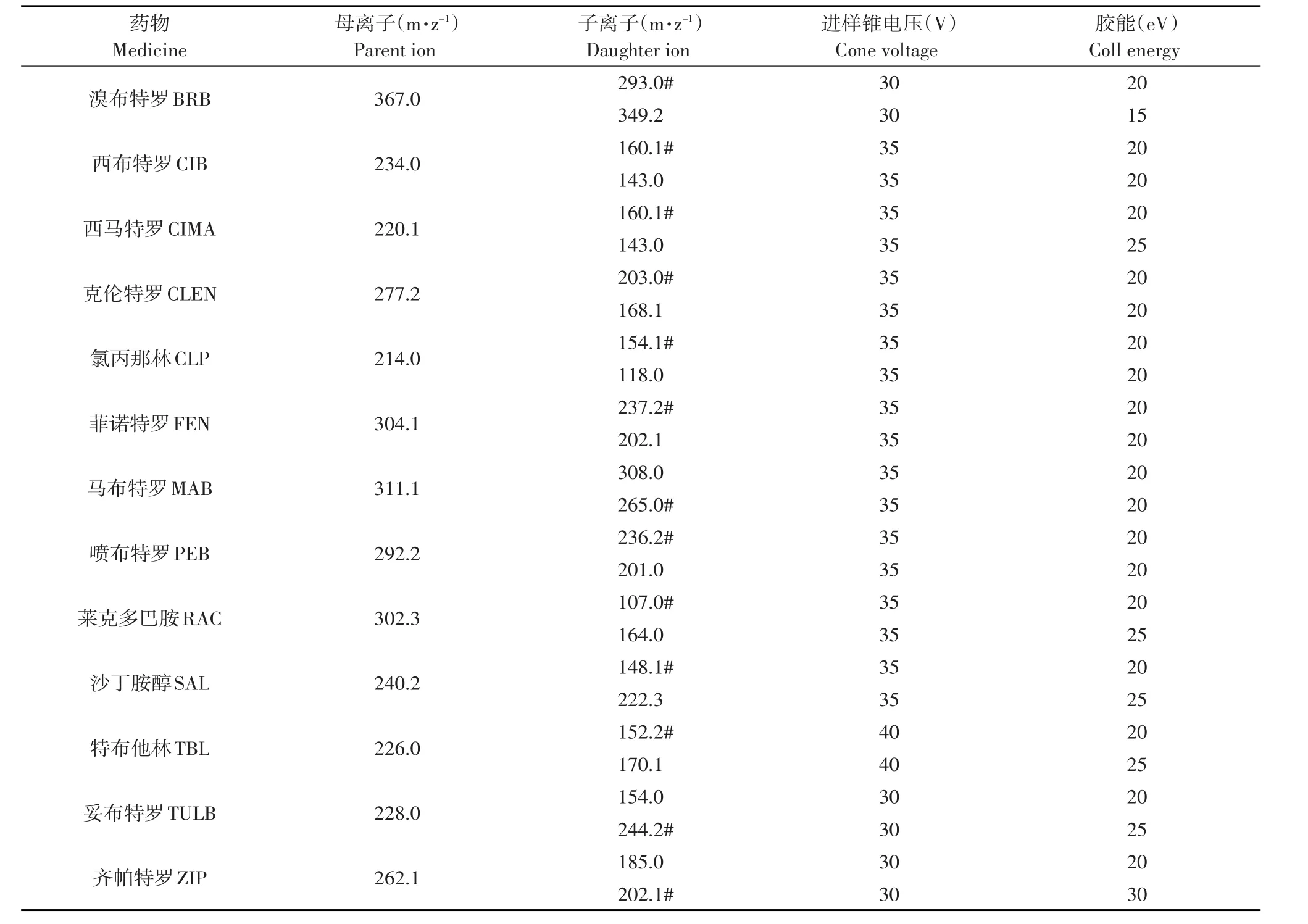
表2 UPLC-MS/MS法测定电喷雾离子源设置Table 2Set of electrospray ion source in UPLC-MS/MS
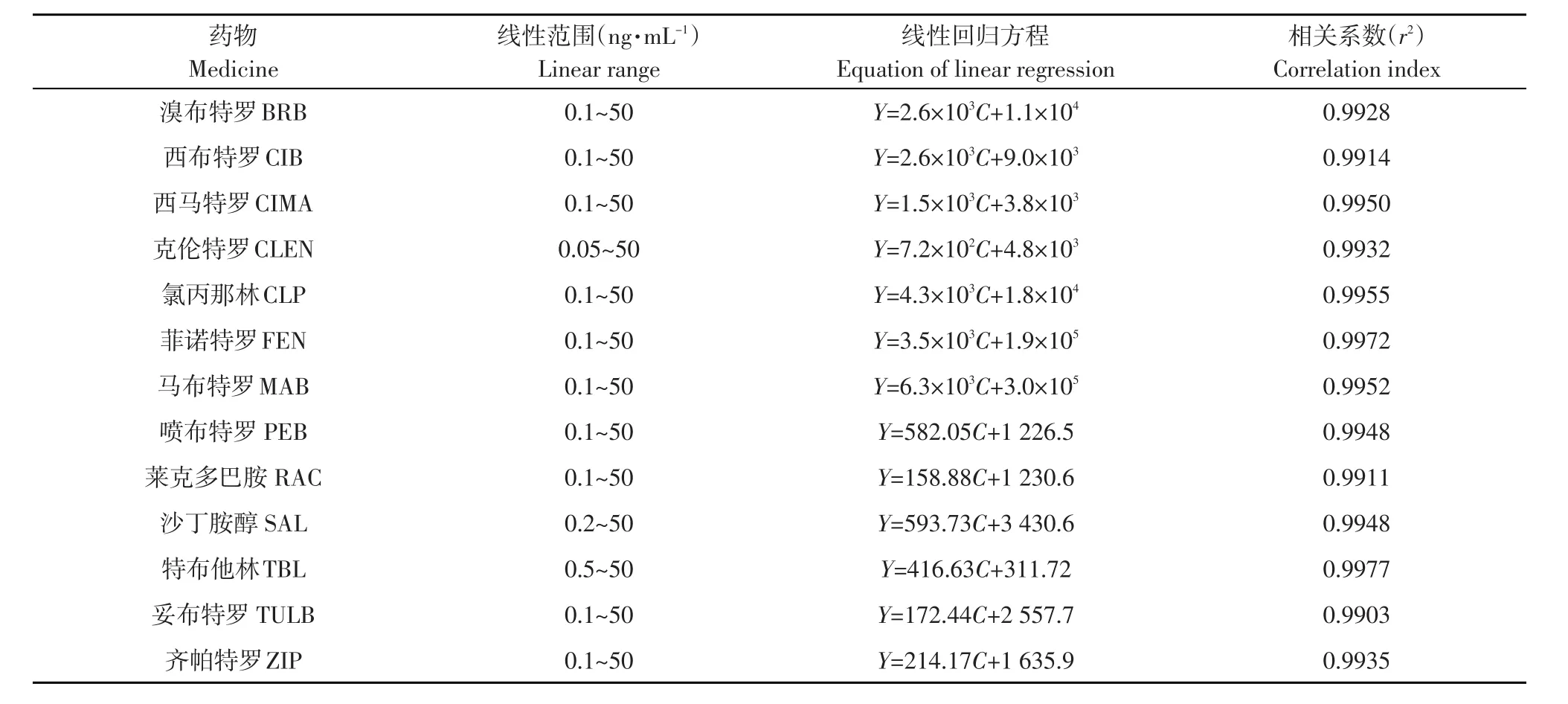
表3 UPLC-MS/MS法测定猪肉中β-受体激动剂类药物线性回归方程及相关系数Table 3Standard curve equations and correlative coefficients of UPLC-MS/MS analysis of β-agonist drugs in animal derived food
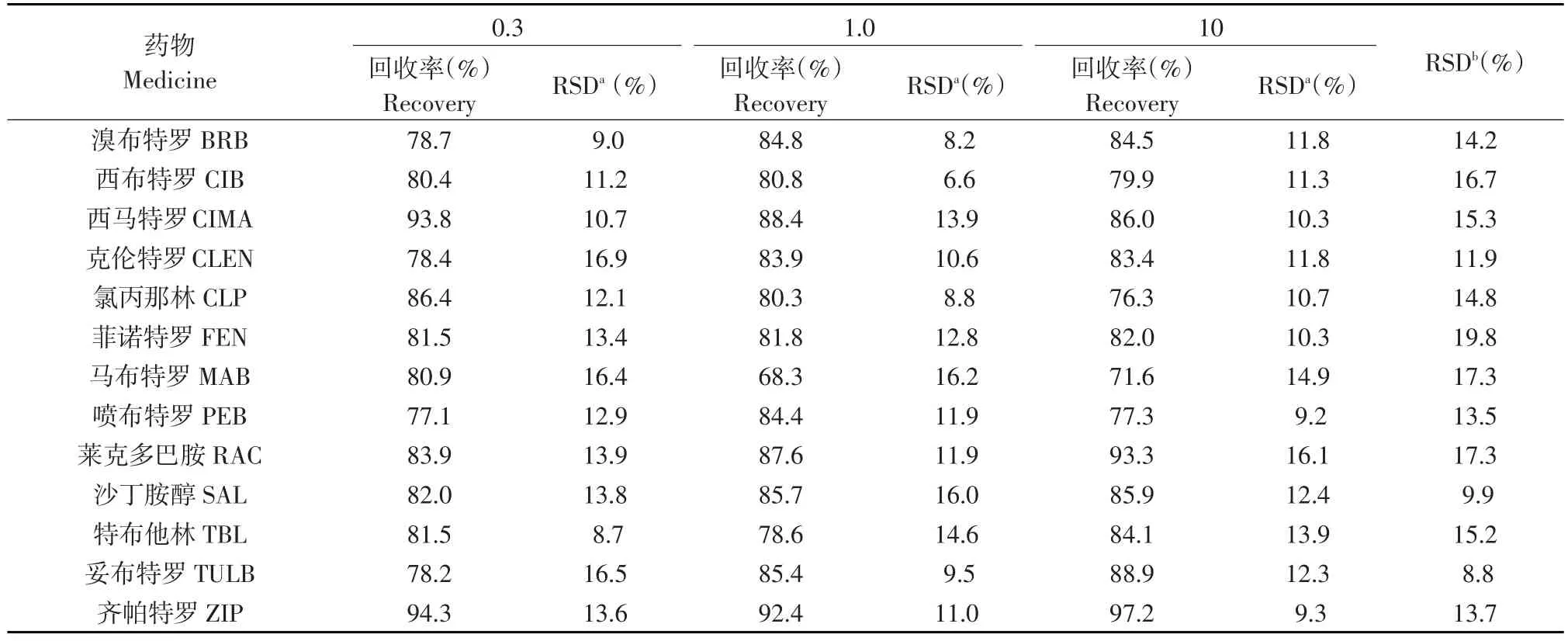
表4 动物肌肉中药物的回收率和日内变异系数(UPLC-MS/MS,n=6)Table 4Drugs in animal muscle recovery rate and coefficient of variation of one of these days(ng·mL-1)
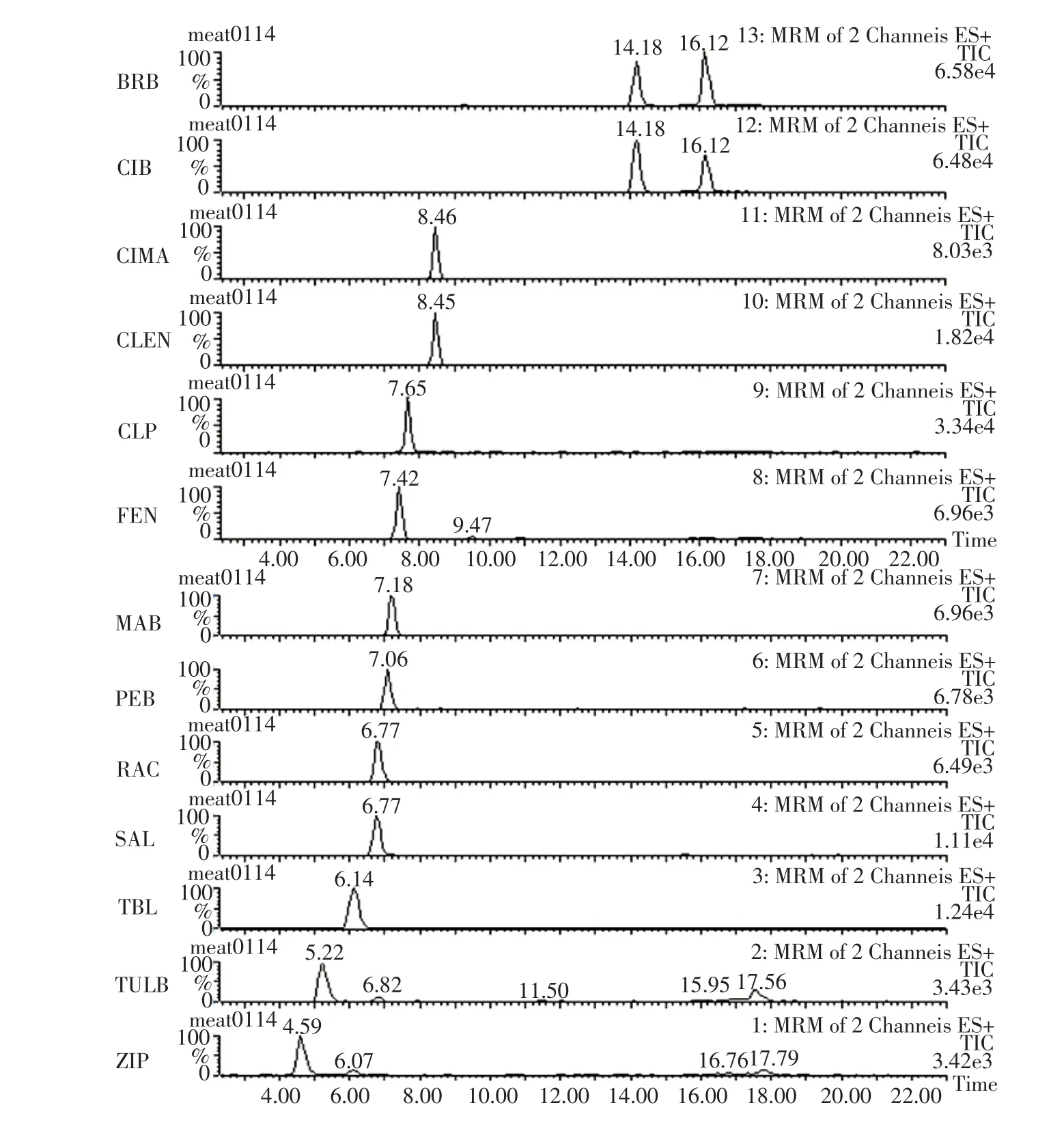
图1 肌肉组织中添加10 ng·mL-1样品UPLC-MS/MS检测总离子色谱图Fig.1Total ion chromatograms of 10 ng·mL-1sample in muscle tissue in UPLC/MS/MS
2.4方法的应用
2013年6月~2014年6月,在黑龙江省哈尔滨市及周边地区采集猪肉类样品共计202份,应用本试验建立的UPLC-MS/MS方法检测13种β受体激动剂类药物,同时作阴性和阳性对照试验,全部样品未检出β受体激动剂类药物(见表5)。
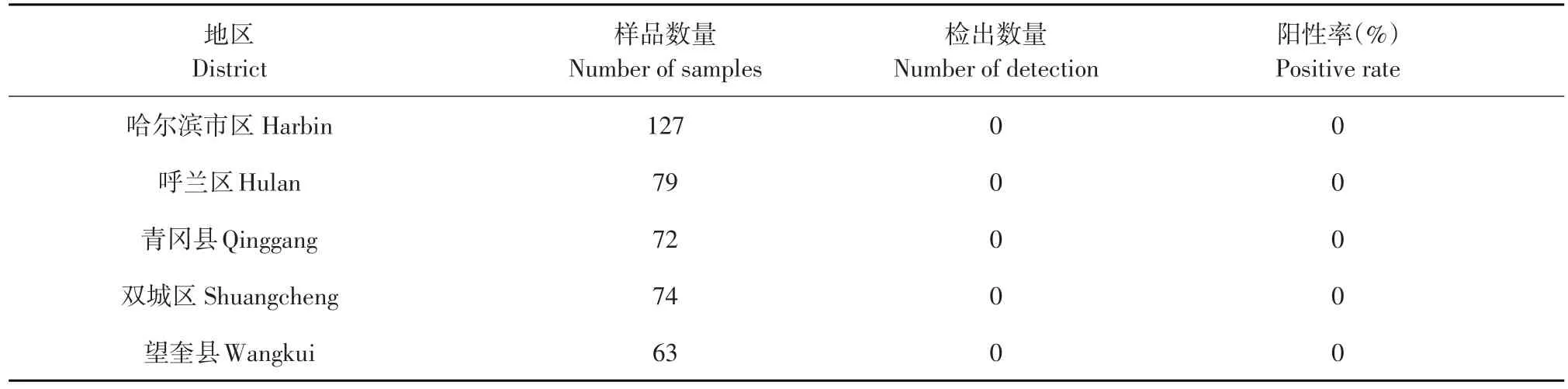
表5 2013年6月~2014年6月期间猪肉中13种β受体激动剂的测定结果Table 5Result of 13 kinds of β-agonists in pork samples from Jun.,2013 to Jun.,2014
3 讨论
3.1检测样品酶解、提取及检测方法对检测结果影响
样品检测方法的稳定性、准确性和灵敏性是关键指标。样品中标记物浓度变化与测得的响应信号应成正比,在一定范围内呈线性关系。浓度与信号之间相关系数R值越大,误差越小,样品检测的稳定性和准确性越好。在色谱测定中,R值应大于0.99。同时,检测样品的酶解和提取是影响色谱测定法的重要环节[12],并且检测方法对检测结果也有很大影响。
β受体激动剂类药物(克伦特罗除外)在动物体内代谢过程中,药物的极性基团容易和体内其他组织成分形成结合物,如与葡萄糖醛苷酸、硫酸酯蛋白发生轭合反应,形成各种轭合物,如尿液中约85%菲诺特罗以葡糖苷酸酶化-硫酸酯蛋白化结合物的形式存在[13],动物肝脏中约40%~45%的沙丁胺醇以结合物形式存在[14]。这些β受体激动剂结合态的存在影响检测结果的准确性;试验表明当样品经过1 000单位β-盐酸葡萄糖醛苷酶-芳基硫酸酯酶,37℃条件下水解16 h可达到理想检测效果[14]。聂建荣等为节省检测时间未采取酶解处理过程[15],而只用高氯酸溶液酸解及加热超声处理沉淀蛋白质杂质,经萃取净化后利用HPLC-MS/MS方法检测动物尿液中β-受体激动剂,检测结果在0.25~20 μg·L-1的浓度范围内线性关系良好,检测定量限为0.25 μg·L-1;且在5.4 μg·L-1浓度的克伦特罗阳性样品5次重复检测分别为4.8、5.4、5.2、5.7和5.0 μg·L-1,平均值为5.2 μg·L-1。孙涛等利用UPLC-MS/MS方法检测猪尿样品时[2],样品酶解处理后,9种β受体激动剂类药物在1.0~100 ng· mL-1浓度范围内呈良好线性关系,检测限值在0.037~0.14 ng·mL-1之间,检测准确性和稳定性更好。采用原有的GC/MC检测方法,虽然样品经包括酶解在内所有前处理,但检测定量限值在0.54~2.50 μg·kg-1水平[16]。本文建立UPLC-MS/MS检测方法在样品处理中,采用β-盐酸葡萄糖醛苷酶-芳基硫酸酯酶在pH 5.2环境中,16 h水解,使药物完全释放,检测结果在0.1~50.0 ng·mL-1浓度范围内具有良好线性关系,R值均大于0.99;且检测限值均为0.1 ng·mL-1,定量限值为0.3 ng·mL-1,检测效果较好。同时,样品中存在高浓度盐分和大分子质量蛋白质,将对色谱峰的峰形、保留时间、分离效果和重现性造成严重影响,故检测前需提纯样品,包括提纯剂沉淀离心处理、液相/固相提取等过程。提纯时使用的提纯剂一般有琥珀酸盐、Na2EDTA-Mcllvaine缓冲溶液和高氯酸等[5],将检测样品pH调节至1.0左右,蛋白质发生变性而不溶于水,β受体激动剂则以相应盐的形式溶于水,离心分离结合有机溶剂萃取法可除去绝大多数杂质蛋白。本试验选用的提纯剂是强酸高氯酸,先将检测样品pH调节至1.0离心除去大多数杂质后,再用氢氧化钠溶液调节pH至9.5后开始液相和固相萃取,结果表明检测数据的准确性和稳定性较好。其他相关检测中采取此类方法,可有效提高检测结果准确性[5-6]。
3.2UPLC-MS/MS检测法应用
利用本文建立的UPLC-MS/MS检测法检测13种β受体激动剂类药物残留,同时设立阳性和阴性对照,全部样品β受体激动剂类药物均呈阴性。为更好地检验本检测方法的准确性和灵敏性,并监督检测临床生产中β受体激动剂类药物的违规使用情况,应进一步扩大临床采样范围和数量以验证检测方法并获得更加准确、全面信息。
4 结论
通过提取条件、净化条件以及仪器分析条件的选择优化,成功建立同时测定猪肉中13种β受体激动剂残留量的UPLC-MS/MS方法,灵敏度、精确度均满足动物源性食品中兽药残留分析要求。
[1]孙雷,张骊,朱永林,等.猪肝中β-受体激动剂残留能力验证结果分析[J].中国兽医杂志,2011,45:41-44.
[2]孙涛,刘圣红,乔昆云,等.超高效液相色谱-串联质谱法测定猪尿中9种β-受体激动剂残留[J].分析仪器,2012(3):48-51.
[3]Pou K,Ong H,Adam A,et al.Combined immunoextraction approach coupled to a chemiluminescence enzyme immunoassay forthedeterminationofenzymeimmunoassayforthe determination of trace levels of salbutamol and clenbuterol in tissue samples[J].Analyst,1994,119(12):2659-2662.
[4]刘畅,吴小虎,徐伟东,等.LC-MS/MS测定动物源性食品中15种β-受体激动剂残留的研究[J].药物分析杂志,2008,28(12): 2085-2089.
[5]M-P Montrade,B Le Bizec.β2-agonistic drugs in urine of meat-producing animals by gas chromatography-mass spectrometry[J].Anal Chim Acta,1993,275:253-268.
[6]Lin LA,Tomlinson J A,Satzger R D.Detection of clenbuterol in bovine retinal tissue by high-performance liquid chromatography with electrochemical detection[J].J Chromatogr A,1997,762(1/ 2):275-280.
[7]Batjoens P,Courtheyn D,De Brabander H F,et al.Gas chromatographic-tandem mass spectro-metric analysis of clenbuterol residues in faeces[J].J Chromatogr A,1996,750(1/2):133-139.
[8]Gonzalez P,Fente C A,Franco C,et al.Determination of residues of the β-Agonist clenbuterol in liver of medicated farm animals by gas chromatography-mass spectrometry using diphasic dialysis as an extraction procedure[J].J Chromatogr B Biomed Appl,1997, 693(2):321-326.
[9]Sangiogi E,Curatolo M.Application of a sequential analytical procedure for the detection of β-agonist brombuterol in bovine urine samples[J].J Chromatogr B Biomed Appl,1997,693(2): 468-478.
[10]Hogendoorn E A,van Zoo nen P,Polettini A,et al.Hyphenation ofcoupled-columnliquidchromatographyandthermospray tandemmassspectrometryfortherapiddeterminationof β-agonist residues in bovine urine using direct large-volume sample injection[J].J Mass Spectrom,1996,31(4):418-426.
[11]Collins S,O'eeffe M,Smyth MR.Multi-residue analysis for β-agonists in urine and liver samples using mixed phase columns with determination by radioimmunoassay[J].Analyst,1994,119 (12):2671-2674.
[12]刁彩霞.禽用抗病灵口服液的血浆药代动力学与安全性实验[D].哈尔滨:东北农业大学,2007.
[13]Pou K,Ong H,Adam A,et al.Combined immunoextraction approach coupled to a chemiluminescence enzyme immunoassay for the determination of enzyme immunoassay for the determination of trace levels of salbutamol and clenbuterol in tissue samples[J].Analyst,1994,119(12):2659-2662.
[14]Doerge D R,Churchw ell M I,Holder C L,et al.Detection and confirmation of β-Agonists in bovine retina using LC/APCI-MS [J].Anal Chem,1996,68(11):1918-1923.
[15]聂建荣,朱铭立,连槿,等.高效液相色谱-串联质谱法检测动物尿中15种β-受体激动剂[J].色谱,2010,28(8):759-764.
[16]朱坚,李波,方晓明,等.气相色谱-质谱法测定肝、肾和肉中11种β-受体激动剂残留量[J].质谱学报,2005,26(3):129-137.
Establishment and application of UPLC-MS/MS method detecting β-agonist drugs in pork meat
LI Guangxing1,HAN Xi1,2,WU Yan3,YOU Xuan2, WANG Junlian2,WEI Jing4(1.School of Veterinary Medicine,Northeast Agricultural University, Harbin 150030,China;2.Heilongjiang Entry-exit Inspection and Quarantine Bureau,Harbin 150090,China;3.Heilongjiang Inspection and Quarantine Bureau Technical Centre,Harbin 150090,China;4.Dalian Entry-exit Inspection and Quarantine Bureau,Dalian Liaoning 116001, China)
This paper established the ultra-performance liquid chromatography tandem mass spectrum(UPLC-MA/MS)method and applied to detect 13 kinds ofβ-agonists in pork samples. β-agonist sample was hydrolyzed by hydrochloride glucose aldehyde glycosides enzyme/aryl sulfatase, purified by mixed cationic exchange(MCX)solid-phase extraction column,analyzed with 0.1%formic acid aqueous solution,0.1%formic acid,and methanol as mobile phase,detected by UPLC-MS/MS. There were 0.3,1.0,10 ng·mL-1of concentration of the added levels in reference pigs muscle tissue samples,the average recovery ratio of 13 kinds of drugs ranged between 68.3%and 97.2%,the variation coefficient in one day ranged between 6.6%and 16.9%,and the variation coefficient of day-to-day ranged between 8.8%and 23.3%.With this detection method,the assay limit of medicine residue is 0.1 ng·mL-1,the optimal detection limit is 0.3 ng·mL-1.Totally 202 pork samples werecollected from Heilongjiang province,13 kinds ofβ-agonists were assayed with the established method and no positive results was observed.
β-agonist,medicine residue,ultra-performance liquid chromatography tandem mass spectrum
S859.84
A
1005-9369(2015)12-0045-07
2015-03-03
国家自然科学基金项目(31172295,31272569)
李广兴(1968-),男,教授,博士,博士生导师,研究方向为动物病理学。E-mail:ligx@neau.edu.cn
猜你喜欢
中成药(2018年10期)2018-10-26
祝您健康(2018年9期)2018-09-04
天然产物研究与开发(2018年6期)2018-07-09
哈尔滨医药(2016年3期)2016-12-01
癌变·畸变·突变(2016年3期)2016-02-27
中国卫生标准管理(2015年25期)2016-01-14
湖南中医药大学学报(2015年1期)2016-01-06
医学研究杂志(2015年2期)2015-06-10
医学研究杂志(2015年5期)2015-06-10
中国卫生标准管理(2015年15期)2015-01-26
