
间质性蕈样肉芽肿1例并文献复习
2015-12-13 05:35秦国敬张福仁陈声利卢宪梅
中国麻风皮肤病杂志 2015年3期
秦国敬张福仁陈声利卢宪梅∗
·论著·
间质性蕈样肉芽肿1例并文献复习
秦国敬1,2张福仁1陈声利1卢宪梅1∗
报道1例间质性蕈样肉芽肿并对相关文献进行复习。患者,男,67岁。全身泛发浸润性红斑伴剧烈瘙痒半年。皮肤科检查:躯干、臀部及四肢见片状红斑,上覆少量白色鳞屑,边界欠清,部分皮损呈浸润性。皮损组织病理学检查示:表皮散在少量淋巴样细胞移入,部分移入细胞见核周空晕及小的Pautrier微脓肿。真皮浅层淋巴样细胞苔藓样浸润。真皮中深部胶原纤维间及血管周围单一核细胞、组织细胞及少许嗜酸粒细胞呈间质性浸润,胶原纤维束间隙增宽,并见黏液物质沉积。浸润的淋巴细胞异型不显著,但CD4、CD3、CD2和CD43均阳性表达。
蕈样肉芽肿; 间质性蕈样肉芽肿
间质性蕈样肉芽肿(Interstitial mycosis fungoides,IMF)是蕈样肉芽肿中罕见的组织学亚型,由Ackerman首先提出,Shapiro等1将此病报道。目前全球共报道本病共13例,2-8其中国内报道2例。为提高对间质性蕈样肉芽肿的认识及诊断水平,现将我们近期诊断的IMF 1例报道如下,并对本病进行了文献复习。
1 临床资料
患者,男,67岁。因全身泛发红斑伴剧烈瘙痒半年来我院就诊。患者半年前无明显诱因于躯干部出现红斑,伴瘙痒,于当地医院就诊,拟诊“过敏性皮炎”,给予注射“门冬氨酸钙注射液”1.5 g每日1次及口服“甲泼尼龙片”80 mg/d治疗,1周后瘙痒明显减轻,皮疹部分好转。患者停服“甲泼尼龙片”,自行外用糖皮质激素软膏,病情反复发作。近期皮疹加重,瘙痒剧烈,来我院就诊。患者既往体健,否认服药史、家族遗传病史及类似疾病史。
体检:一般情况可,全身浅表淋巴结未触及肿大。心肺腹检查未见异常。皮肤科检查:躯干、臀部及四肢见片状红斑,上覆少量白色鳞屑,边界欠清,部分皮损呈浸润性(图1a,b)。腰背部及双胫前呈鱼鳞病样改变(图1c)。皮损约占体表面积的60%。临床拟诊为蕈样肉芽肿。
实验室检查:血尿常规、乙肝五项、肝肾功能、电解质、血细胞沉降率及C反应蛋白未见明显异常;肿瘤标记物检查阴性;心电图正常;肝、胆、胰、脾B超检查示:脂肪肝、胆囊结石;浅表淋巴结B超检查未见明显肿大;胸部X线检查未见异常。
腹部红斑组织病理学检查示:表皮灶性角化过度及角化不全,棘层轻度增厚,个别淋巴细胞移入表皮。真皮浅中层血管周围及胶原间淋巴样细胞呈间质性浸润,个别细胞核大深染,胶原间见黏液物质增多。组织病理主要表现为间质性炎症改变,未见典型的
MF改变。随选取右下肢浸润性皮损组织病理学检查示:表皮轻度角化过度,轻度棘层增生,散在少量淋巴样细胞移入,移入淋巴样细胞见核周空晕及Pautrier微脓肿形成(图2a)。真皮浅层胶原增生,淋巴样细胞呈苔藓样浸润。真皮中深部胶原纤维间及血管周围淋巴细胞、组织细胞及嗜酸粒细胞呈间质性浸润(图2b)。胶原纤维束间隙增宽,可见黏液物质沉积。阿新蓝染色:胶原束间较多黏液物质沉积(图2c)。浸润细胞轻度异型。免疫组化染色:CD4(图3a,b)、CD43、CD3、CD2均阳性表达,CD8约20%阳性,CD56约20%阳性,MPO小于10%阳性,Ki-67约30%阳性,CD20、CD79a(图3c)、CD30均阴性。CD68散在组织样细胞阳性。诊断:间质性蕈样肉芽肿(斑片/斑块期)。
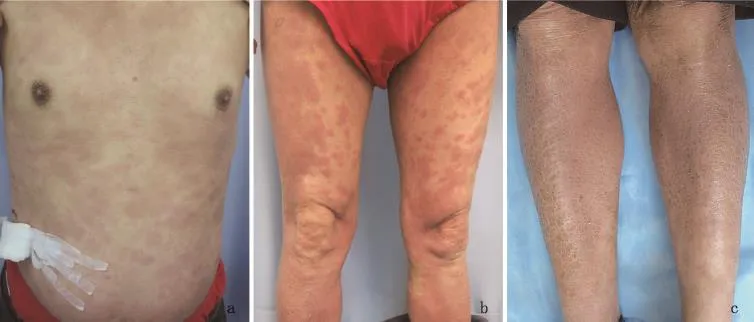
图1 a:胸、腹部见片状红斑,上覆少许白色鳞屑,边界欠清,部分皮损呈浸润性;b:双下肢见片状红斑,边界欠清,部分皮损呈浸润性;c:双胫前呈鱼鳞病样改变

图2 a:(右下肢浸润性皮损)表皮轻度角化过度,轻度棘层增生,散在少量单一核细胞移入,部分移入细胞见核周空晕及Pautrier微脓肿(HE,×100);b:真皮中深部胶原间散在淋巴样细胞,细胞核异型不著(HE,×200);c:胶原间黏液物质沉积,胶原纤维似漂浮于黏液湖中(阿新兰染色,×400)

图3 a:表皮内及真皮内浸润细胞CD4阳性(SP,×100);b:真皮胶原束间浸润细胞CD4阳性(SP,×100);c:真皮浅层、真皮胶原束间及移入表皮的单一核细胞均不表达CD79a(SP,×100)
治疗:给予口服阿维A胶囊10 mg每日3次,雷公藤多苷2片每日3次。随访2个月,躯干、四肢皮损基本消退,臀部未见明显改善。目前仍在随访中。
2 讨论
蕈样肉芽肿(Mycosis fungoides,MF)是最常见的原发性皮肤T细胞淋巴瘤,其临床及组织学亚型多样。2005年世界卫生组织(WHO)和欧洲癌症研究与治疗组织(EORTC)共同制定皮肤淋巴瘤分类方法(WHO-EORTC皮肤淋巴瘤分类法),该分类方法将MF分为经典的MF和3种MF变异亚型,即亲毛囊性MF,Paget病样网状细胞增多症和肉芽肿性皮肤松弛。除此之外还有间质性MF、亲汗管性MF、丘疹型MF等,但由于病例数少、仅临床表现或组织病理学单方面有特点,而没有单独列出。现结合本例对IMF进行简要总结。
2.1 临床表现 IMF连同本例国内外文献报道共12例(排除2例以法语报道的病例),平均年龄63.17± 13.02岁,36~77岁,其中男性4例,女性8例。MF多发于中老年人,国外文献报道平均确诊年龄为55~60岁,7-10国内报道平均确诊年龄为44.2~50.5岁。11可看出IMF患者平均年龄较国内外统计的平均年龄偏大。文献报道MF患者男∶女比例约为1.6~2.0∶1,7-10而IMF患者男:女比例为1∶2,似乎多见于女性。
文献报道的12例患者均表现为红斑、斑块伴少量鳞屑,主要位于臀部、躯干及四肢,其中1例还累及头皮,21例仅发生于右腰背部,5其余10例表现为躯干及四肢均受累,11例患者表现为多发性皮损。其中6例伴不同程度的瘙痒。因此,IMF临床表现并无特异性,大部分为多发性皮损表现,主要发生于MF早期,2但也有文献报道主要发生于斑块期或肿瘤期。12
2.2 组织病理 9例患者组织病理出现不同程度的异型淋巴细胞亲表皮现象,6例出现真皮浅层带状淋巴细胞浸润和真皮胶原间黏液物质的增多。免疫组化主要表现为单克隆辅助性T细胞表型。TCR单克隆基因重排仅有2例表现为阳性。2
所以IMF组织病理学主要表现为真皮浅层淋巴细胞带状浸润及亲表皮现象,可见Pautrier微脓肿;真皮中下部胶原纤维束间明显的淋巴样细胞、组织细胞浸润,胶原束间隙增宽,间有黏液样物质沉积。Ferrara等3报道,可以出现胶原纤维漂浮于黏液湖中的现象,即free-floating collagen fibers。并且移入表皮与真皮胶原束间的淋巴样细胞均表现为相同的单克隆辅助性T细胞表型。但也有患者主要表现为淋巴细胞呈间质性浸润,亲表皮现象及异型不明显,此时诊断会非常困难,临床与病理密切结合则变得非常重要。
此外,有7例患者(含本例)胶原纤维束间出现较多CD68+细胞,2,3因此有学者认为IMF和肉芽肿性MF是宿主对真皮浸润肿瘤细胞的免疫反应的不同表现方式。
2.3 鉴别诊断 由于IMF病理主要呈间质性浸润模式,临床又多见于MF的斑片期及斑块期,易造成诊断上的困难。根据本病的诊断过程及文献复习,需与伴有间质性浸润的皮肤病相鉴别。4①间质性环状肉芽肿,当未发展为丘疹或环状时,皮损表现相似,但一般无瘙痒等自觉症状。组织病理学上无亲表皮现象、Pautrier微脓肿及真皮乳头纤维增生伴苔藓样浸润等MF的表现,而且间质中浸润的细胞主要为CD68+的组织细胞,有时见多核巨细胞。②伴较多炎症细胞的局限性硬皮病,组织学为多克隆的淋巴细胞、浆细胞及组织细胞在间质内浸润,伴程度不等的胶原纤维增生,无真表皮的界面改变,且临床表现不同。③间质性肉芽肿性药物反应,起病一般较急、且起病前有用药史,停用致敏药物皮损可消失。组织病理学表现为累及真皮间质和血管周围的弥漫性肉芽肿性皮炎,浸润细胞主要为组织细胞、淋巴细胞、嗜酸粒细胞、浆细胞、上皮样细胞及多核巨细胞,常伴巨细胞吞噬胶原纤维及弹性纤维碎片。④间质性肉芽肿性皮炎,常是结缔组织病等系统疾病的皮肤表现,皮损特征多为丘疹和结节,常呈条索状、弧形、环状和斑块状。组织病理学表现为以组织细胞为主的间质性浸润,小片状胶原坏死灶及其周围较多嗜中性粒细胞和嗜酸粒细胞围绕;多核巨细胞和黏蛋白沉积无或很少见到。免疫组化染色主要是CD68强阳性,而淋巴细胞标记及Ki-67均为阴性。⑤白血病累及皮肤,有白血病病史,组织学上具有髓过氧化酶(MPO)等骨髓源性标记的淋巴样细胞在胶原间浸润,结合血液细胞学和骨髓象等,可与本病鉴别。由于MF临床及病理表现的多样性,临床详细查体及多部位、多次取材病理检查对于明确诊断是必要的。
本例患者临床表现符合MF,腹部斑片期损害组织病理学主要表现为淋巴细胞间质性浸润,缺乏典型的亲表皮及淋巴细胞的异型,造成诊断上的困难。重取具有浸润性皮损,组织学发现表皮及真皮浅层具备了典型的MF组织学模式,而真皮中深部仍为淋巴细胞、组织细胞间质性浸润模式,并伴较多黏液物质沉积。免疫组化显示大部分浸润的淋巴细胞为单克隆辅助性T细胞表型,因此符合间质性MF改变。由于浸润的细胞达真皮深部,结合临床部分皮损具明显的浸润感,病期应在斑片/斑块期。
2.4 治疗 IMF由于报道较少,现无标准治疗方案。由于亲毛囊性或亲汗管性MF浸润较深,因此通常联合用PUVA和维A酸类药物。13本例患者考虑到浸润达真皮深部,给予口服阿维A胶囊和雷公藤多苷等也
取得较好的疗效。此外,Targretin可用于各期MF,可以单用,更多时候是联合其他治疗。在文献复习中发现患者采用氮芥软膏(浓度为10%~40%)、PUVA、Targretin、电子束等治疗可获得较好的效果。
由于本病报道较少,其生物学行为、治疗、预后等还需进一步研究。
1 Shapiro PE,Pinto FJ.The histologic spectrum of mycosis fungoides/Sézary syndrome(cutaneous T-cell lymphoma).A review of 222 biopies,including newly described patterns and the earliest pathologic changes.Am J Surg Pathol,1994,18(7):645-667.
2 Su LD,Kim YH,LeBoit PE,et al.Interstitial mycosis fungoides,a variant of mycosis fungoides resembling granuloma annulare and inflammatory morphea.J Cutan Pathol,2002,29(3): 135-141.
3 Ferrara G,Crisman G,Zalaudek I,et al.Free-floating collagen fibers in interstitial mycosis fungoides.Am J Dermatopathol,2011,32(4):352-356.
4陈浩,薛燕宁,万川,等.间质性蕈样肉芽肿一例.中华皮肤科杂志,2011,44(12):851-853.
5陈思远,杨珍,夏颖,等.单一损害的间质性蕈样肉芽肿一例.中华皮肤科杂志,2013,46(4):270-272.
6 Paoletti MT,Comozb F,Dompmartin-Blancherec A,et al.Interstitial mycosis fungoid:a rare variant of mycosis fungoids. Two cases.Ann Pathol,2011,31(1):36-40.
7 van Doorn R,Van Haselen CW,van Voorst Vader PC,et al. Mycosis fungoides:disease evolution and prognosis of 309 Dutch patients.Archives Dermatol,2000,136(4):504-510.
8 Zackheim HS,Amin S,Kashani-Sabet M,et al.Prognosis in cutaneous T-cell lymphoma by skin stage:long-term survival in 489 patients.J American Academy Dermatol,1999,40(3):418-425.
9 Kim YH,Liu HL,Mraz-Gernhard S,et al.Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression.Arch Dermatol,2003,139(7):857-866.
10 Wain EM,Orchard GE,Whittaker SJ,et al.Outcome in 34 patients with juvenile-onset mycosis fungoides.Cancer,2003,98(10):2282-2290.
11刘洁,王宝玺,渠涛,等.51例蕈样肉芽肿临床特征.中国医学科学院学报,2007,29(2):174-180.
12 Cerroni L,Gatter K,Kerl H.Skin Lymphoma:The Illustrated Guide,3rd edn.Oxford:Blackwell Science Ltd,2009,11-46.
13 Leverkus M,Rose C,Bröcker EB,et al.Follicular cutaneous T-cell lymphoma:beneficial effect of isotretinoin for persisting cysts and comedones.Br J Dermatol,2005,152:193-194.
(收稿:2014-11-30 修回:2015-01-02)
Interstitial mycosis fungoides:a case report and review of the literature
QIN Guo-jing,ZHANG Fu-ren,CHEN Sheng-li,et al.Shandong Provincial Institute of Dermatology and Venereology,Provincial Academy of Medical Science,Jinan,250022
A patient with interstitial mycosis fungoides was reported and the relevant literature was reviewed.A 67-year-old male patient presented with a 6-month history of infiltrated erythematous patch with sever pruritus all over the body.Dermatological examination revealed ill-defined light pink macules with white lamellar scales located on the trunk and buttocks.Some of the lesions were infiltrated.Histopathological examination revealed a small number of mononuclear cell infiltrate in the epidermis,with perinuclear halo in some cells and formation of Pautrier's microabscesses.There were lichenoid infiltrates in the superficial dermis and infiltrates consisting of numerous mononuclear cells,histiocytes and a few eosinophils between collagen fibers and around the vessels in the middle and deep dermis.The clefts between collagen fibers were broadened,where mucin deposits were seen.There was no cytologic atypia in the lymphoid cells and immunohistochemically the cells were positive for CD4,CD3,CD2 and CD43 staining in the superficial and middle dermis.
mycosis fungoides;interstitial mycosis fungoides
1山东省皮肤病性病防治研究所,济南,250022 2济南大学山东省医学科学院医学与生命科学学院,济南,250062
∗通信作者
猜你喜欢
人人健康(2022年13期)2022-11-25
西部皮革(2022年19期)2022-11-20
西部皮革(2022年21期)2022-11-16
中国病理生理杂志(2022年6期)2022-07-06
中国典型病例大全(2022年9期)2022-04-19
全科医学临床与教育(2021年4期)2021-05-11
昆明医科大学学报(2020年12期)2021-01-26
云南医药(2020年5期)2020-10-27
中国麻风皮肤病杂志(2019年8期)2019-08-23
天津医科大学学报(2019年3期)2019-08-13
