
傅里叶变换离子回旋共振质谱仪:过去、现在与未来*
2015-12-07 08:32李树奇鲍晓迪殷红孔祥蕾
大学化学 2015年4期
李树奇 鲍晓迪 殷红 孔祥蕾
(南开大学元素有机化学国家重点实验室 天津 300071)
今日化学
傅里叶变换离子回旋共振质谱仪:过去、现在与未来*
李树奇 鲍晓迪 殷红 孔祥蕾**
(南开大学元素有机化学国家重点实验室 天津 300071)
对傅里叶变换离子回旋共振高分辨率质谱仪的发展历程、串联质谱解离技术及最新的应用进行了详细描述,并对其发展趋势进行了展望。
傅里叶变换质谱 离子回旋共振 高分辨率质谱 串联质谱 超导磁体
1974年,Comisarow和Marshall首次将傅里叶变换技术应用到离子回旋共振质谱中,发明了傅里叶变换离子回旋共振质谱仪(Fourier transform ion cyclotron resonance mass spectrometer,FT ICR MS) 技术。在过去的40年中,这项质谱分析技术得到了快速的发展。它集超高分辨率,超高质量精度和高灵敏度等优良性能于一体,在蛋白质组学,生物医药,石油组学,材料科学等众多领域有着非常广泛的应用。另一方面,近年来各种新型离子化方法的发明以及串联质谱新技术的引入,为FT ICR MS开辟了更为广阔的应用空间。
FT ICR MS作为质谱仪器中的贵族,不仅是因为其超高分辨率与精确度,也是指仪器本身高昂的售价,以及定期维护导致的高运行成本。因此,虽然商业化的FT ICR MS早在20世纪80年代就已经出现,但其早期在我国的推广非常有限。然而近15年来情况有了很大的改变,一方面国内用户的数量逐年增多;另一方面其应用的领域也得到很大的扩展。尽管如此,国内用户对于FT ICR MS的认识主要集中于其应用方面,而对仪器本身的发展历程、新方法的融入以及功能扩展等方面的知识却鲜有介绍。与已有的相关综述文章[1-4]不同的是:本文从FT ICR MS仪器本身和其技术发展的角度,对其发展历史、串联质谱解离技术及最新的应用进行了详细描述,并对其进一步发展的前景进行了展望和评述。
1 FT ICR MS的历史与发展
要更好地了解FT ICR MS的发展史,必须要追溯到之前就已经发明的回旋共振质谱。下面我们分别就其整个发展史中的标志性事件进行必要的回顾和说明。
1.1 回旋加速器与回旋共振质谱
1.1.1 粒子回旋加速器(1930)
回旋运动是离子在FT ICR MS分析室中的重要运动形式,其运动方程可以表示为ωc=qB/m。其中ωc为离子回旋运动的角频率,B为磁场强度,q为离子的电量,m为离子的质量。首先提出这一理论的是美国伯克利加州大学物理学教授劳伦斯(Ernest Lawrence)[5],其灵感来源于直线加速器加速工作原理,当直线加速器加速带电粒子时,粒子是沿着一条近于直线的轨道运动被逐级加速的;因此,在需要很高的能量时,加速器的直线距离会很长。1930年,劳伦斯提出了回旋加速器的理论,他做了两个结构相当简单的加速器模型,把氢离子注入后,显示出了使离子回旋加速的效果。1931年,他和他的学生利文斯顿(M. S. Livingston)一起,研制了世界上第一台回旋加速器,利用2kV的加速电压使氘离子加速到80keV的能量,向人们证实了他们所提出的回旋加速器原理。1932年又建成了一台直径为24cm的较大回旋加速器,其被加速粒子的能量可达到1MeV[6]。由于回旋加速器方面的贡献,劳伦斯获得了1939年诺贝尔物理学奖。劳伦斯理论表明,离子在恒定磁场中回旋运动的频率与离子的质量成线性关系,当外加横向交变电场的频率与离子回旋的频率相等时,离子能够被激发到更大的轨道半径。这一发现的意义不仅在于利用较小的电场使带电粒子获得高的能量,也为后来FT ICR MS的发展迈出了重要的第一步。
1.1.2 Omegatron质谱(1949)
离子回旋质谱最早可追溯到1949年设计的Omegatron质谱[7-8]。利用回旋运动原理,Hipple,Sommer和Thomas设计了一种用来准确测量法拉第常数的装置,其设计如图1a 所示,通过轴向的电子束(平行于磁场方向)在分析室区产生离子,以一个捕获电压产生的电场来防止离子在轴向溢出;在两平行电极板上施加与磁场方向垂直的射频(RF)电压,调节RF的频率,当所提供的RF频率与离子的回旋频率相等时,离子吸收能量被加速,以螺旋形轨道行进;在适当的位置放置一个离子收集器,当离子被加速到轨道半径为1.0cm时撞击离子收集器产生共振信号,而那些没有被共振激发的离子则不能被收集。因为该装置测量的是频率ω,他们将其命名为Omegatron。该仪器捕获离子的能力很有限,质量分辨率低,因此一直未能作为一种常规分析质谱,其主要用途是气体分析和泄漏检测。
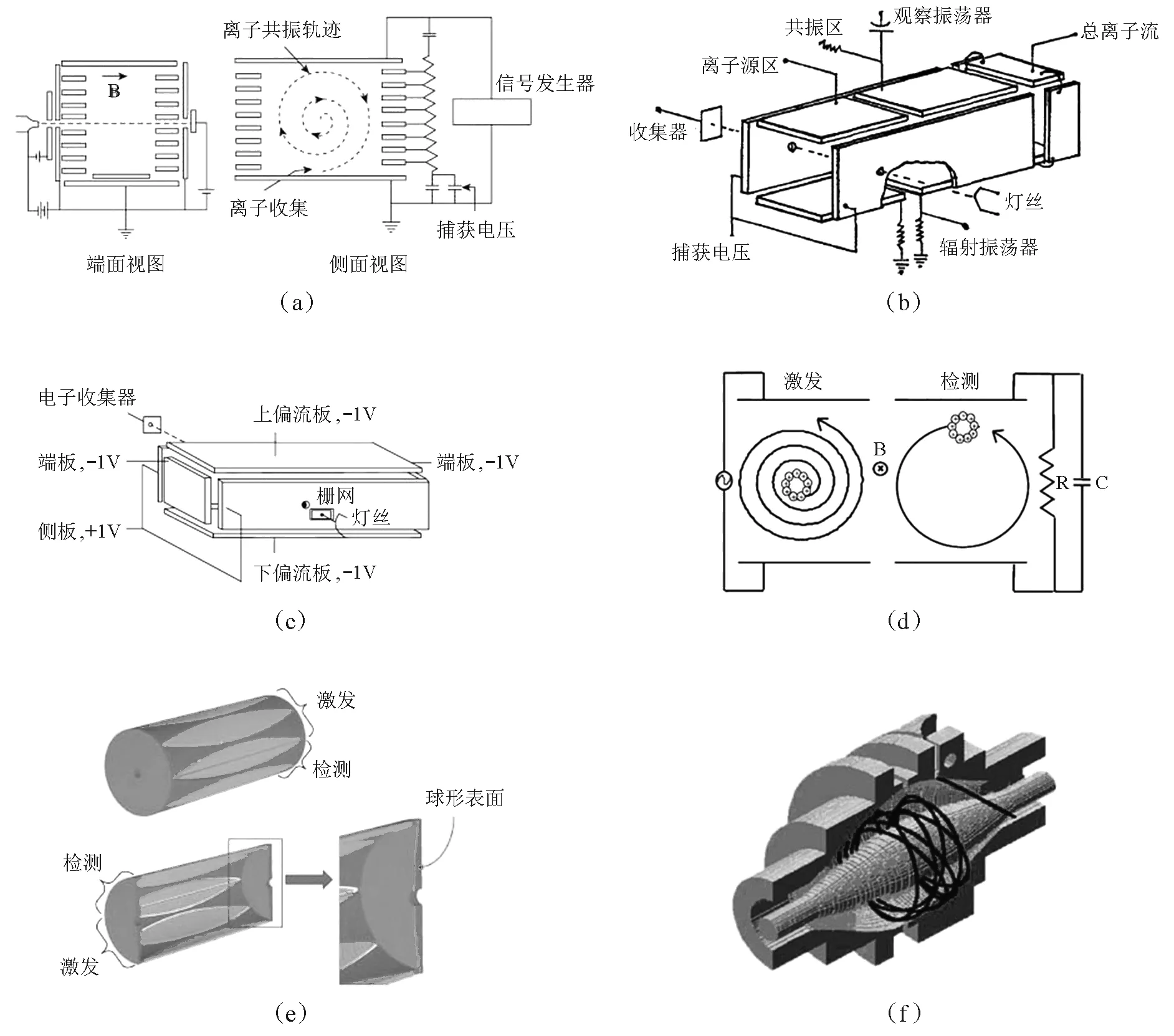
图1 离子回旋质谱 、傅里叶变换离子回旋共振质谱仪以及轨道阱技术 (a) Omegatron结构简图[8] ;(b) 三区结构ICR分析室[9];(c) 捕陷离子型分析室[10]; (d) FT ICR MS的激发和检测过程[11];(e) 和谐阱技术[12];(f) 轨道阱技术[13]
1.1.3 离子回旋共振质谱(1963)
利用ICR原理发展成真正意义上的质谱是在20世纪60年代中后期,在此期间产生了漂移室型和捕陷离子室型两种ICR质谱。漂移室型最早出现于1963年,分析室由离子源区和漂移区组成[14],后来发展为如图1b所示的三区结构:离子源区、共振区和离子收集区[9]。采用电子轰击(EI)的办法在离子源区中产生离子,电子束的方向平行于磁场方向。然后在与电场和磁场垂直的方向施加很小的漂移电压,将离子引入共振区中。为避免电子束对观察振荡器的影响,共振区和离子源区分开引入漂移电压。在磁场和静电场的作用下,离子在与其垂直的方向上沿摆线轨道漂移,为防止离子沿磁场方向逸出, 在两个边面板上加一个小的捕陷电压。当时检测激发回旋运动使用的是既能产生回旋激发需要的射频电场又能测量功率吸收的边缘振荡器。当固定边缘振荡器频率时,扫描磁场强度使样品离子的回旋频率依次与边缘振荡器的频率相等,测量功率吸收随磁场强度的变化来获得样品的质谱图。
1970年,McIver展示了单区捕陷离子室型质谱[10](图1c),其分析室为6个金属极板组成的长方体构型。与三区结构分析室不同,它在垂直于磁场方向多出两个端板作为捕集板,在其上加与离子同极性的小电压就可以形成一个电势阱,将离子囚禁于其中。这种构型的特点是:(1) 离子的产生和检测都在同一区室中进行;(2) 离子能够被较长时间捕获在分析室中(由过去的最长1~2毫秒提高到了100毫秒);(3) 离子从产生到检测再到清除的整个过程通过时间脉冲序列来控制。由于采用脉冲式激发,所有的离子都在同一时间内生成,这就避免了以往电子束的空间电荷对离子检测的干扰。同时,脉冲序列概念的提出对后来傅里叶变换技术在ICR MS上的应用以及发展多级质谱在时间串联上的实现具有重要意义。此后,ICR MS成为气相离子化学领域,尤其是研究离子-分子反应的有效工具。不过采用固定频率激发,磁场扫描的检测模式导致ICR分析速度很慢,另外分析室的真空压力一般控制在1.3×10-5Pa,使仪器的分辨率和质量检测范围都受到限制,这就严重制约了其在分析领域中应用的扩展。
1.2 傅里叶变换质谱
1.2.1 傅里叶变换离子回旋共振质谱 (1974)
ICR MS的飞跃发展是在1974年Marshall和Comisarow成功地将傅里叶变换技术应用到其中之后[15-16]。早在1970年以前,FT技术就被应用到了红外光谱仪和核磁共振波谱仪(NMR)[17]。对于NMR,核的共振频率ω只决定于旋磁比γ与外加磁场强度B,这与ICR的ωc只受限于离子的荷质比q/m和外加磁场强度B相类似,这就给ICR MS工作者以启发。不过ICR与FT技术结合面临几个关键技术问题,主要为:(1) 需要合适的方法将所有离子较长时间囚禁在分析室中;(2) 需要合适的方法对所有离子共振激发;(3) 需要合适方法对所有离子进行同时检测;(4) 与NMR相比,FT ICR MS的数据量更为庞大,需要足够大的计算机内存来支持傅里叶变换对于数据的处理。
McIver的单区捕陷离子型分析室可以实现对离子的较长时间囚禁。对于离子的激发,Marshall和Comisarow采用了快速频率扫描的模式。在较短的时间内,在激发板上引入涵盖了所有离子回旋频率的宽频域射频信号(chirp),在此信号的激发下,相同质量的离子会粘合到一起形成离子团,所有离子都激发到较大半径做回旋运动。对于信号的检测,与过去扫描磁场监视边缘振荡器功率吸收的模式完全不同,他们直接检测离子团在接收板上感应出的镜像电流,该电流包含了所有离子信息的时间域瞬态信号(自由衰减信号),通过宽带RC电路将其放大存储为电压信号后,利用傅里叶变换进一步转化成频率域信号,进而得到以质荷比为横坐标的质谱图。图1d描述了离子团被激发和检测的过程,这种宽带激发和检测的模式大大提高了分析效率,也显著改善了仪器的分辨率,并扩展了它的质量范围。目前,这种模式仍然是FT ICR常规测试中最常用的激发和检测方法。1975年,Marshall等用这种仪器获得了m/q为1885的三全氟代庚基吖嗪的分子峰,在相对分子质量为28范围内获得了分辨率为250000的CO、N2和C2H4混合物的超高分辨谱,还获得了质量范围为69~605的全氟煤油质谱,显示出该种仪器在分析应用方面的巨大潜力[18]。1978年,他们搭建了第一台FT ICR MS[19],从此傅里叶变换高分辨质谱技术被确立成为研究复杂离子化学的强有力分析方法[11]。
1.2.2 世界首台商业化FT ICR MS(1981)
1981年,Nicolet仪器公司推出了世界首台商业化FT ICR MS,型号为FT MS-1000(图2a),最初采用磁场强度为1.2特斯拉(T)的电磁体,后来又升级为1.9T和3.0T的超导磁体。1982年,Bruker CMS-47 FT ICR MS也加入到市场竞争中来,其磁体为4.7T的超导磁体。FT ICR仪器的质量分辨率和扫描速度与磁场强度B成正比,质量精确度、动态范围与质量检测上限与B2成正比;此外超导磁体更加稳定,因此提高磁场强度是提高该类仪器综合性能的最简单也是最有效方式。商品仪器的出现,尤其是超导磁体的使用以及计算机的飞速发展,推动了FT ICR的研究和应用。作为一个对比,图2b显示了2013年Bruker公司展示推出的新型FT ICR MS(型号为SolariX XR)。

图2 世界首台与目前商业化FT ICR MS仪器 (a) Nicolet FTMS-1000(1981);(b) Bruker SolariX XR(2013)
1.3 新电离源的引入——电喷雾与基质辅助激光解吸电离(1989)
现在,FT ICR MS已经几乎可以和各种电离技术联用。但在早期的仪器中,离子源均为内离子源,对于不易挥发的样品,如聚合物和各种生物样品等,一般把样品点在进样杆上靠近分析室进行电离。这种方法容易对分析室造成污染,此外也影响了分析室的真空度进而降低分辨率。如果要将离子从外离子源引入到分析室中,就得克服超导磁体边沿磁场对离子的反射。离子传导技术的出现,成功解决了这一问题,可以有效地将外离子源产生的离子注入到分析室中。快原子轰击(FAB)以及后来出现的电喷雾(ESI)[20]和基质辅助激光解吸电离(MALDI)[21-22]都属于外离子源电离方法。这些新型的电离方法与FT ICR MS的结合使其很快由传统的气相离子化学的基础性研究领域扩展到有机物及生物分子的结构分析新领域。
用ESI电离技术对生物大分子进行质谱分析最早出现于1988年,第二年它就被成功地应用于FT ICR MS中[23]。ESI属于软电离技术,特别适合对极性分子及热不稳定的分子进行分析。它的最具魅力的特点就是能够形成多电荷离子,而且所带的电荷数随着相对分子质量的增加而增加。对于ICR,由回旋运动公式可以看出相对分子质量越大的离子其回旋运动频率越小,这就使得其检测大分子离子较为困难。ESI电离技术的出现,使得大分子离子带多个电荷出现在FT ICR可检测频率区域,而其特有的超高分辨能力可以将多电荷的同位素峰簇分辨开来确定其具体的电荷数,进而精确测定出大分子物质的相对分子质量。此外,多电荷电离可以提供多个电荷并产生很强的镜像电流,从而可以提高FT ICR的灵敏度,甚至可以检测到带有很多电荷的单个离子[24]。1988年,Tanaka,Hillenkamp分别提出使用基质辅助来获得大分子离子[21-22],该方法将样品加入到能强烈吸收激光的基质中,基质将激光的能量传递给样品,以产生分子离子,从而避免了直接用激光照射分析物,为分析生物大分子提供了一个理想的方法。MALDI的特点是能耐受适量的缓冲剂和盐,灵敏度很高,容易出现单电荷离子,碎片离子较少。MALDI最早是在1991年与FT ICR实现联用[25],它与ESI是相互补充的质谱技术,它们与FT ICR的联用被广泛用于生物样品、高聚物等大分子及石油、中草药等复杂体系的研究。尽管其他类型的质量分析器也可以实现与MALDI、ESI的联用,但FT ICR特有的超高分辨率与精确度,无疑使其在成分确认和结构分析上更具优势。
1.4 新型分析池(2011)
历史上曾出现过多种几何形状的分析池(cell),目前广泛使用圆柱形cell,因为此种cell能够更好与磁体中央的圆洞(bore)匹配,增加空间体积,减少离子空间电荷效应。但在离子检测时,cell两端的捕集电压形成的电场会影响仪器的分辨率。为此,在离子检测时一般采用尽可能小的捕集电压。另外一种方法则是创建一个双曲电场,使得离子回旋运动的频率与它们的轴向运动无关。然而,早期方法所提供的双曲电场仅仅集中在cell中心很小的空间内[26-27]。2011年,Nikolaev等在理论分析与数字模拟的基础上,设计并实现了一种新型FT ICR cell (被称之为“和谐阱”)[12,28]。该圆柱形分析池表面被均分成多个独立单元,这些独立单元内部产生同等大小的双曲电势场,将相隔单元并联组成两组电路,分别作为激发电极和探测电极,它们与捕集电极一起构成整个分析池(图1e)。该方法大大提高了仪器分辨率。用此新型cell在7T FT ICR上对利血平(reserpine,质子化分子离子峰为609)进行测试,获得了高达2400万的超高分辨率;对牛血清白蛋白(BSA),在宽带模式下获得了电荷数从37到60的多电荷谱图分布,在窄带模式下对电荷数为49的同位素峰簇(m/z1357)进行测试,获得了该峰簇的清晰分布,其分辨率高达120万。分辨率的极大提高,也使得这种新型cell测试的质量上限超过了10万[28]。
2 FT ICR MS中使用的串联质谱技术
分析池是FT ICR的核心部件,它集质量分析器和离子检测器于一体,利用不同的时间段施加不同的脉冲事件完成激发和检测。由于FT ICR采用时间串联的方式工作,离子检测不是以牺牲离子为代价,因此可以对检测后的离子做多种多级质谱研究。20世纪90年代后,FT ICR技术发展的一个重要方面就是多种解离技术的出现与发展。迄今,与FT ICR联用的串联质谱技术主要有碰撞诱导解离、红外多光子解离、黑体红外辐射解离、表面诱导解离和电子捕获解离。
2.1 碰撞诱导解离(collision-induced dissociation,CID)
CID(有时亦被称为碰撞活化解离,CAD)是用在FT ICR上的一种传统解离技术[29]。被隔离的母离子被尽可能低的捕获电压囚禁于分析池中,在这种情况下,离子不会产生任何碎片。只有将离子激发到更大半径并与惰性气体(如N2)发生碰撞才能获得较大的能量。离子接受能量后发生分子内振动能量的重新分布,如果提供的能量高于要断裂键的键能,离子裂解。CID主要断裂肽键,产生以b和y类型为主的离子峰。
有多种方法可以增加离子动能,最为常用的是共振激发和持续非共振辐射(sustained off-resonance irradiation,SORI)[30]。在共振激发中,用与离子回旋频率完全一致的RF脉冲信号将离子激发到较大半径,该脉冲信号的强度与持续时间决定了离子的最后动能。该方法常用来研究相对较小的分子在单次碰撞后裂解的碎片信息。如果发生多次碰撞,离子的动能会急剧衰减,随后碰撞的效率会很低而不利于大分子离子的裂解。在SORI中,母离子受到一个稍微偏离其共振频率的低强度RF信号激发,使其回旋半径不断膨胀或收缩。由于RF强度很低,离子不会偏离分析池中心太远。在激发时,通过脉冲阀引入碰撞气体增大分析池中的气压。在这种情况下,离子的碰撞概率大大增加,尽管每次的碰撞仅仅有一小部分能量被转换(通常≤0.3eV),但是经历千百次这样的碰撞,母离子最终累积达到足够多的内能而裂解。由于内能是逐步积累升高的,它随机遍布于全部离子当中,因此母离子以低能碎裂途径裂解。当碎片产生后,它们的回旋频率不再与RF产生偏共振,也就不会被加速与氮气发生碰撞,而会慢慢冷却下来变得更稳定。因此,子离子一旦产生就会冷却下来,不会继续裂解下去。
2.2 红外多光子解离(infrared multiphoton dissociation,IRMPD)
IRMPD最初用来对小分子进行化学研究,后来扩展到对生物大分子进行结构表征[31]。IRMPD与SORI-CID相似,只是能量供给方式不同。IRMPD多采用CO2红外激光器。将激光器置于磁体的末端,激光被激发后就会通过磁体的孔洞照射到ICR分析池。离子对光子的多次吸收产生内能的积累,同SORI一样,IRMPD给离子所提供的能量也是缓慢和随机的,离子的分解经低能过程进行(对于10.6μm波长的CO2红外激光的光子能量为0.12eV)。与SORI不同的是,碎片离子经SORI-CID产生后,在氮气中很快冷却,不会进一步分解;而在IRMPD过程中,如果激光一直打开的话,则碎片离子将继续吸收光子积累内能进一步发生低内能的裂解,从而使产生的解离谱的碎片信息更为丰富。此外,IRMPD过程是在分析池中心进行的,不需要脉冲事件对离子进行激发,使仪器采集速度加快;同时由于没有类似CID实验碰撞气体的引入,使分析池的高真空状态得以维持,从而避免了瞬态信号快速衰减导致的分辨率降低。
2.3 表面诱导解离(surface induced dissociation,SID)
SID是用固体表面作为碰撞靶,选定的母离子被加速与其碰撞后动能转变为内能而解离,生成的产物离子从表面散射出去,随后由检测器将其检出。对于不同类型的碰撞表面,采用含硅或碳氟化合物的涂布材料的表面解离效率最好,而石墨表面和碳水化合物表面的内能转化率则较低。此外,内能的转化率还和离子束与碰撞表面的角度有关。SID传递给母离子的内能分布较窄,易于控制内能的转化。当选定的母离子与表面碰撞以后会产生多种反应,这些反应所占的比例与碰撞表面的性质和碰撞的角度有关。若要得到离子的结构信息,应当选择发生非弹性碰撞[32]。此外,离子与表面碰撞会发生电荷交换导致母离子中性化,该反应不仅使母离子的电荷转移到表面上,同时会产生涂布材料的小分子离子,影响低质量范围离子分析。SID首次应用于FT ICR是在1990年[33-34],离子进入阱后再被加速至阱壁或加速至置于分析池后捕获集端的内置探头,发生碰撞。与CID类似,它们都是使母离子中化学键较弱的位置先发生断裂,容易形成b和y类型为主的碎片离子。不同的是,SID不需要将碰撞气引入到分析池当中,从而减小了对真空系统的破坏。
2.4 黑体红外辐射解离(blackbody infrared radioactive dissociation,BIRD)
BIRD是从热解离技术中发展起来的、主要应用于FT ICR质谱仪器的气相离子解离技术[35-36]。它与IRMPD技术相类似,也是通过低能红外光子的多次吸收来解离,它们的区别主要在于辐射源不同。BIRD是利用热电偶探测温度变化,对处在高真空条件下的分析池进行加热,池壁产生黑体红外光子。BIRD实验的温度变化范围一般是从室温到约250℃,是一个慢热过程,解离反应一般会持续几秒甚至几分钟。在BIRD实验之前经常会利用短脉冲的惰性浴气体,与选定的母离子发生碰撞对母离子冷却,以免其与背景中的中性分子发生随机碰撞导致碰撞诱导解离发生。对于大分子离子,在BIRD实验初期,离子与池壁辐射的红外光子不断进行着能量的交换,内能得到积累,此时离子吸收和辐射光子的速率要远大于解离速率,离子的内能按照玻尔兹曼分布规律达到平衡。当该诱导期结束后,母离子的内能通常会呈现指数式衰减,同时相应的碎片离子产生。离子的解离速率及裂解途径与分析池温度直接相关。相比其他解离技术,BIRD技术更容易获得精确的解离能和离子解离的动态信息。
2.5 电子捕获解离(electron capture dissociation,ECD)
ECD是由McLafferty小组在1998年发明的针对FT ICR质谱的裂解技术,主要用于多肽和蛋白质的分析[37-38]。它通常是将电喷雾所产生的带有多电荷的离子引入到FT ICR分析池中,离子与分析池后的阴极发射装置发射的电子(一般<1eV)相遇,电子被正离子捕获,离子发生裂解。与CID、IRMPD和BIRD等断裂总是发生在能量最低的化学键产生b、y系列离子不同,ECD是由低能量的自由电子与质子化的多电荷离子在相互作用的过程中由于放热而瞬间产生碎裂,它是一个非各态历经(non-ergodic)的过程,断裂反应先于分子内振动能量重新分布进行,主要产生由N—Cα键的断裂而形成的c、z系列离子。ECD技术适合于氨基酸序列长度较长的、带3个或3个以上电荷为主的肽段解离。它对蛋白质和多肽的主链断裂没有歧视,裂解覆盖程度高,翻译后修饰基团不会丢失,优先断裂二硫键。ECD的这些技术优势与以往解离方法在数据上有很好的互补性,在对蛋白翻译后修饰研究和利用“Top down”策略对蛋白质序列分析方面有着广阔的应用前景。不过,ECD不能使非共价键断开,这就阻碍了其对高相对分子质量蛋白的断裂,尤其是对于20kDa以上的蛋白质,常规ECD的断裂作用就微乎其微了。为了突破该限制,对常规ECD技术进行了改进,如活化离子ECD技术,在ECD前使用氮气脉冲(或红外照射、黑体辐射等)对大相对分子质量的母离子进行碰撞,使离子在电子捕获前高级结构被破坏。采用红外照射活化ECD方法已经实现了对45kDa蛋白质的鉴定[38]。另外,单电荷母离子捕获电子后会中和而不显电性,无法使用ECD分析。此外,灵敏度不高也是ECD技术需要改进的方面。
3 近年来FT ICR MS在生物领域中的应用
3.1 在蛋白质组学研究中的应用
高性能FT ICR MS能够准确测定蛋白质中多肽链的数目和各肽段的氨基酸序列,有效提高鉴定的速度和可信度;与多维色谱联用能够高效精准地进行鸟枪法蛋白质组学研究;还能够用Top down途径分析蛋白质翻译后修饰。而如今,在蛋白质组学中,质谱技术更适合于高通量的蛋白组测定,快速完整地获得蛋白质的氨基酸信息是生物质谱发展的方向。Ge Ying等用电子捕获解离技术(ECD)成功地对大肠杆菌的细胞抽提物中催化维生素B1生物合成的几种酶进行了二硫键的全面结构鉴定,此技术提高了蛋白质组学中大的蛋白质的细节结构鉴定[39]。2014年,Lakshmanan等在液相色谱联用FT ICR MS上采用独立数据碎片方法对自上而下的蛋白质水解酶蛋白进行识别[40]。另外,研究蛋白质与其他生命物质的复合物也具有重要意义。Nousiainen等以SORI-CID和电喷雾FT ICR MS研究了钙调蛋白RS20-Ca4复合物的气相解离,一定程度上揭示了复合物中非共价相互作用的主要来源[41]。
3.2 在代谢组学以及中药现代化研究中的应用
代谢组学是通过比较对照组和实验组的代谢组(metabolomes,某一生物的所有代谢物组分),以寻找代谢谱差异的研究,是研究生物体内的小分子代谢物质变化规律的科学。它是功能基因组学和系统生物学研究的重要组成部分,也是生命科学内继基因组学和蛋白质组学之后新发展起来的一种组学。普遍认为,如果能把蛋白质组学、转录组学和代谢组学的数据整合在一起,将能破译基因的功能和阐明基因的活动规律,也就能获得生命有机体生物学更完整的图像。对代谢组学而言,傅里叶变换质谱由于其高分辨率和高准确度等特点,可以不采用色谱分离,便能直接进行代谢物的检测和鉴定。2012年,Konstantinos等利用FT ICR MS和气相色谱的联用,研究出关于马铃薯的病原体入侵时的一套完整的代谢网络[42]。2014年,Sadakatali等利用FT ICR质谱图研究了硫醇的代谢物并量化分析了细胞谷胱甘肽[43]。
另一方面,高分辨质谱由于具有质量范围宽、扫描速度快、灵敏度高的优点,可提供母离子和碎片离子的精确质量数及可能的元素组成,因此在中药化学成分的快速鉴定中得到广泛的应用。在中药及其复方的定性和定量研究中,高分辨质谱起着重要的作用,其分析结果为全面了解中药及其复方的有效成分和药理学的研究奠定了基础。刘春明等[44]利用FT ICR MS高分辨质谱以及其他一些技术手段,对朝鲜淫羊藿中生物碱类进行分析及鉴定,并发现了一种新的生物碱。2014年,左冉等为了深入研究黄连解毒汤体内发挥疗效的效应物质基础,用液相色谱联用FT ICR MS方法发现并确证了给予黄连解毒汤后大鼠血浆中主要的38个入血成分[45]。随着FT ICR MS的广泛应用于中药的分析和鉴定,必将为我国的传统医学事业做出巨大的贡献,也将有利于推动中医药向世界的进一步推广。
4 展望
首台FT ICR MS问世至今已40年。随着电子技术、软件技术以及离子化方法的发展,近年来FT ICR在性能高端化、易操作化、多功能化等方面有了很大的发展。同时,生命科学的快速发展对高分辨质谱又提出了更新、更高的要求。展望未来,FT ICR MS仍有较大的发展空间,主要体现在以下几个方面。
4.1 对超导磁体提出了更高的要求
超导磁体是FT ICR的核心部件。由于更高的磁场强度不仅会大幅度提高其质谱分辨率和降低测量误差,而且也能扩展其测量范围。追求超高磁场来适应其在相关科研领域的发展是其最重要的一个发展方向。当前大多数实验室FT ICR配备的超导磁体为7T、9.4T 或 12T、14.5T 与15T高性能仪器目前已经在一些实验室开始使用,21T(相当于核磁900M)系统目前已经建成。 但另一方面,超导磁体的高额成本及维护成本却限制了其在各个领域,尤其是作为常规分析仪器推广。其中一个非常强的竞争对手即为Orbitrap质谱仪[13,46]。Orbitrap采用静电场轨道阱作为离子回旋振荡的质量分析器(图1f),它通过探测离子的镜像电流,并将时间域信号转化为频率域信号,进而得到质谱图。尽管在分辨率等指标方面,Orbitrap稍逊一些,但是其价格相对便宜,体积小了很多,使用和维护也更为方便。Orbitrap作为一个轻量级的FT MS,可以满足大多数高分辨率质谱实验的要求,已受到越来越多的用户青睐。为面对这一挑战,FT ICR MS除了在进一步提高自身的高端品质外,也更加重视采用新技术来降低其运行成本。一个典型的例子就是目前FT ICR的超导磁体一般多采用一个超低温泵将挥发出来的气体氦重新压缩成液氦注入到磁体当中,以减少液氦的充装次数,提高仪器运行的经济性和环保性。
4.2 更丰富的串联质谱功能
现在的FT ICR仪器除具有传统的时间串联质谱功能(如SORI-CID,IRMPD与ECD等)外,多在离子源后串联一个四级杆质量分析器和一个六级杆碰撞池(quadrupole和hexapole,Qh)或者线性离子阱组成混合型傅里叶变换质谱,使其具有类似于三重四级杆或Q-TOF质谱仪的Qh CID串联质谱功能。利用混合型FT ICR进行二级质谱分析,碎片在Qh区域或线性离子阱内产生,高分辨探测在FT ICR池中进行,这比在分析池中做传统的时间串联分析简单快捷,同时也大大提高了二级质谱的分析速度,解决了其与液相色谱联用兼容性较差的技术难题。此外,杂合Qh可富集低丰度目标离子极大提高了微量关键样品的检测能力。正是因为这一技术革新,使FT ICR解决复杂样品和进行复杂实验的能力大大提高,特别适用于生命科学蛋白质组学的精细研究,扩展了它在生命科学的应用领域。
4.3 更丰富的离子源
目前,FT ICR能够兼容各种类型的离子源并与多种传统或独特的解析方法相结合,可提供最大的实验灵活性。除了ESI与MALDI这两种最常用的离子化方式,它可以选择搭配大气压化学电离(APCI),大气压光电离(APPI)和纳喷雾(nanoESI)等多种电离方式。另外,如Bruker推出的复合离子源,通过电脑控制可以轻松完成ESI与MALDI之间的切换,而且两个源不仅可以单独使用,还可以同时使用,这在为用户提供应用便利的同时也提供了新的实验能力。而近年来发展起来的常压离子化技术,如电喷雾解吸电离技术(DESI)[47],实时直接分析电离技术(DART)[48]和基质辅助激光解吸电喷雾电离技术(MALDESI)[49]等,可将复杂样品直接离子化或者只需对样品做简单的预处理。常压离子化技术与FT ICR联用,为FT ICR对复杂体系样品同时分析提供了新的思路,使其应用领域得到进一步拓展。
4.4 综合性能更强
性能高端化与应用功能多元化是FT ICR发展的一个方向。以Bruker的最新型SolariX XR为例:Apollo Ⅱ离子漏斗电喷雾源能够在宽质量范围提供高的离子传输效率;优化的离子透镜系统提供了更宽质量范围和更高灵敏度;偏轴离子捕集技术减少了离子飞行带来的质量歧视效应,提高了仪器的扫描速度、质量范围和灵敏度;而最新采用的和谐阱技术[28],可提供全范围离子的稳定性和高达数千万的分辨率,可以对化合物同位素精细结构进行深入分析,揭开之前遗漏的信息。因此,此类仪器将可以应用在其它类型质谱难以解决的高端应用领域,包括生物大分子精细分析、小分子组织分子成像、代谢组学分析、石油组学和食品、饮料、尿液、环境等复杂体系分析、未知物定性定量分析等。
[1] 余天,郭寅龙,张尊建,等.分析测试技术与仪器,1999,5(4):202
[2] 王光辉,熊少祥,何美玉,等.现代仪器,2001,1:1
[3] 王伟,蔡文生,邵学广.化学进展,2005,17(2):336
[4] 李明,梁大鹏,李红梅,等.仪器仪表学报,2011,32(8):1915
[5] Lawrence E O,Edlefsen N E.Science,1930,72:376
[6] Lawrence E O,Livingston M S.PhysRev,1932,40:19
[7] Hipple J A,Sommer H,Thomas H A.PhysRev,1949,76:1877
[8] Sommer H,Thomas H A,Hipple J A.PhysRev,1951,82:697
[9] Beauchamp J H,Anders L R,Baldeschwieler J D.JAmChemSoc,1967,89(18):4569
[10] McIver R T Jr.RevSciInstrum,1970,41(4):555
[11] Comisarow M B,Marshall A G.JMassSpectrom,1996,31:581
[12] Boldin I A,Nikolaev E N.RapidCommunMassSpectrom,2011,25:122
[13] Hu Q,Noll R J,Li H,etal.JMassSpectrom,2005,40:430
[14] Wobschall D,Graham J R,Malone D P.PhysRev,1963,131(4):1565
[15] Comisarow M B,Marshall A G.ChemPhysLett,1974,25(2):282
[16] Comisarow M B,Marshall A G.ChemPhysLett,1974,26(4):489
[17] Ernst R R,Anderson W A.RevSciInstrum,1966,37(1):93
[18] Wilkins C L.AnalChem,1978,50:493
[19] Marshall A G,Comisarow M B,Parisod G.JChemPhys,1979,71:4434
[20] Fenn J B,Mann M,Meng C K,etal.Science,1989,246:64
[21] Karas M,Hillenkamp F.AnalChem,1988,60:2299
[22] Tanaka K,Waki H,Ido Y,etal.RapidCommunMassSpectrom,1988,2:151
[23] Henry K D,Williams E R,Wang B H,etal.ProcNatlAcadSciUSA,1989,86(23):9075
[24] Chen R,Wu Q,Mitchell D W,etal.AnalChem,1994,66:3964
[25] Hettich R L,Buchanan M V.JAmSocMassSpectrom,1991,2:22
[26] Bruce J E,Anderson G A,Lin C Y,etal.JMassSpectrom,2000,35:85
[27] Tolmachev A V,Robinson E W,Wu S,etal.JAmSocMassSpectrom,2008,19:586
[28] Nikolaev E N,Boldin I A,Jertz R,etal.JAmSocMassSpectrom,2011,22:1125
[29] Dienes T,Pastor S J,Schurch S,etal.MassSpectromRev,1996,15:163
[30] Gauthier J W,Trautman T R,Jacobson D B.AnalChimActa,1991,246:211
[31] Little D P,Speir J P,Senko M W,etal.AnalChem,1994,66:2809
[32] Dongre A R,Somogyi A,Wysocki V H.JMassSpectrom,1996,31:339
[33] Williams E R,Henry K D,McLafferty F W,etal.JAmSocMassSpectrom,1990,1:413
[34] Ijames C F,Wilkins C L.AnalChem,1990,62:1295
[35] Dunbar R C.MassSpectromRev,2004,23:127
[36] Price W D,Schiner P D,Williams E R.AnalChem,1996,68:859
[37] Zubarev R A,Kelleher N L,McLafferty F W.JAmChemSoc,1998,120:3265
[38] Tsybin Y O,Ramstrom M,Witt M,etal.JMassSpectrom,2004,39(7):719
[39] Ge Y,Lawhorn B G,EINaggar M,etal.JAmChemSoc,2002,124:672
[40] Rajeswari L,Jeremy J W,Rudy A,etal.Proteomics,2014,14:1271
[41] Nousiainen M,Vainiotalo P,Feng X.EurJMassSpec,2001,7(4&5):393
[42] Aliferis K A,Jabaji S.PLoSONE,2012,7(8):e42576.doi:10.1371/journal.pone.0042576
[43] Sadakatali S G,Pawel L,Daniel S E,etal.AnalBioanalChem,2014,406:4371
[44] Liu C M,Liu Z Q,Dou J P.ChemJChinUniv,2003,24(12):2215
[45] Zuo R,Wang H J,Si N,etal.ActaPharmaceuticaSinica,2014,49(2):237
[46] Makarov A.AnalChem,2000,72(6):1156
[48] Cody R B,Laramée J A,Durst H D.AnalChem,2005,77:2297
[49] Sampson J S,Hawkridge A M,Muddiman D C.JAmSocMassSpectrom,2006,17:1712
FourierTransformIonCyclotronResonanceMassSpectrometer:Past,PresentandFuture*
Li Shuqi Bao Xiaodi Yin Hong Kong Xianglei* *
(StateKeyLaboratoryofElemento-OrganicChemistry,NankaiUniversity,Tianjin300071,China)
The history and development of Fourier transform ion cyclotron resonance mass spectrometer (FT ICR MS) is presented in detail. The dissociation technique for tandem mass spectrometry and the latest application are introduced. The development trend of FT ICR MS is discussed.
Fourier transform mass spectrometry; Ion cyclotron resonance; High resolution mass spectrometry; Tandem mass spectrometry; Superconducting magnet
10.3866/pku.DXHX20150401
国家自然科学基金(Nos.21172121,21121002,21475065)
**通讯联系人,E-mail:kongxianglei@nankai.edu.cn
O6;G64
猜你喜欢
食品安全导刊(2021年20期)2021-08-30
国际口腔医学杂志(2019年3期)2019-05-31
天然产物研究与开发(2018年2期)2018-04-04
中学生数理化·高一版(2016年7期)2016-12-07
试题与研究·中考化学(2016年1期)2016-09-30
当代化工研究(2016年5期)2016-03-20
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
医学研究杂志(2015年11期)2015-06-10
特产研究(2014年4期)2014-04-10
中国环境科学(2014年4期)2014-02-02
