
拟南芥基因转录调控关系的计算方法研究
2015-11-17 12:11于晓庆
应用技术学报 2015年1期
于晓庆
(上海应用技术学院理学院,上海 201418)
拟南芥基因转录调控关系的计算方法研究
于晓庆
(上海应用技术学院理学院,上海 201418)
拟南芥是一种重要的模式植物,已被广泛应用于植物生物学研究.基于基因表达谱和序列信息构建了预测拟南芥基因调控关系的数学模型.通过支持向量机和夹克刀的测试,结果表明该方法在拟南芥基因调控关系的预测工作中有很好的表现.利用计算方法预测拟南芥基因转录调控关系可为实验室研究提供一定理论依据.
拟南芥;调控关系;支持向量机;计算方法
生物体的发育过程由一系列复杂的基因调控网络控制,一个完整的基因调控网络由转录因子(transcription factor,TF)、靶基因(target gene,TG)及其之间的调控关系构成.其中,转录因子是基因表达的重要调控因子,在高等生物体的生命循环中起关键性作用,它们结合在其靶基因启动子序列上的特定位点,从而激活或抑制下游靶基因的表达.这些特定的位点,一般是长度为5~25 bp的DNA序列片段,称为转录因子结合位点(transcription factor binding site,TFBS).预测转录因子及其靶基因的调控关系对深入研究基因的调控网络、生物体发育的分子机理具有重要研究意义[1-2].
近20年来,识别转录因子和靶基因调控关系的方法很多,其中最著名的实验方法是Chip-on-chip技术.该技术利用染色质免疫沉淀技术和微阵列技术,能够在试管中识别出一个特定转录因子的启动子.但这种基于实验技术的方法受实验环境及其他因素的限制,且需大量时间和资金.因此,随着生物数据的大量积累和计算工具的发展,利用生物信息学方法预测转录因子和靶基因的调控关系被不断地开发出来.这类方法主要分为两种:一种是利用表达谱信息的相关性,通过建立一定的数学模型预测调控关系;另一种是利用识别转录因子结合位点的方法,寻找一组共表达基因的共motif过程来预测调控关系.目前,对拟南芥这种模式植物,基因调控关系的预测方法主要以实验方法为主,而利用生物信息学手段来预测其调控关系的方法极为少见.本文基于基因的表达谱数据以及序列的信息特征构建调控关系的特征向量数学模型,并利用支持向量机预测拟南芥的基因转录调控关系.
1 材料与方法
1.1 数据来源
拟南芥基因表达谱数据从美国Stanford拟南芥信息资源网站(The Arabidopsis Information Resource,TAIR)下载得到.这些微阵列表达数据来源于拟南芥不同的组织和发展阶段:seeding and whole plant,leaves,roots,flower and pollen,shoots and stems,siliques and seed.同时,在数据库Arabidopsis Gene Regulatory Information Server(AGRIS)中下载了拟南芥转录因子的蛋白质序列,并从拟南芥全基因组中提取了每个基因的上游启动子序列,长度最大为1 000 bp.
1.2 实验方法
拟南芥基因调控关系预测的具体流程如图1所示.
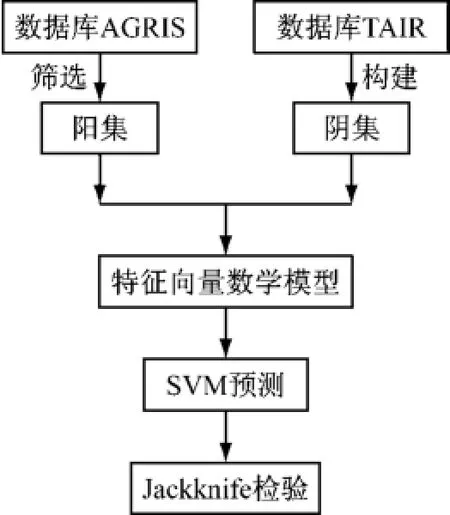
图1 预测模型流程图Fig.1 Flow chat of prediction model
1.2.1 数据集的构建
样本数据包括阳集数据和阴集数据.阳集数据是指经实验证实具有调控关系的基因对.所使用的阳集数据是来自于数据库AGRIS中下载的598对标记为“confirm”的、经实验验证的调控关系基因对筛选后得到的.具体筛选过程为:首先,筛选掉在TAIR数据库中不含有相对应转录因子蛋白质序列或靶基因启动子序列的基因对;其次,去掉基因不存在表达谱数据的基因对;最后,为缓解样本数据的不平衡性,将调控关系多于20的转录因子所对应的基因对随机减少到20.通过以上步骤,最终构建了一个含有156对调控关系的阳集数据集.
阴集数据,是指确定没有基因调控关系的基因对.但目前为止,对于模式植物拟南芥,并没有文献公开发表哪些基因对是确定没有调控关系的.阴集数据在生物数据分类问题中对于分类器的可靠性起着关键性的作用.采用以下策略构建了阴集:对于转录因子TF,若不存在对应的转录因子结合位点TFBS,则随机选择一个基因作为靶基因TG与其构成一个基因对(TF,TG).为确保TG不被转录因子TF所调控,随机重新排列基因TG的表达谱顺序,保证了调控关系的不存在性;若TF含有对应的TFBS,则在所有基因中搜索该TFBS.若基因TG中不含有TF的TFBS,那么(TF,TG)就构成了一个阴集样本.对于拟南芥这种模式植物,实际的阴集和阳集的比例可能达到1 000∶1,甚至更大.通过减少阴集样本数量的方法可以缓解训练样本的不均衡性并提高方法的预测性能.为找出合适的比例参数,比较了不同参数的预测性能,最终选择1∶4作为阳集与阴集的最优比例.构建了624个阴集样本数据,数量是阳集数据的4倍,样本数据情况如表1所示.
1.2.2 特征向量模型的构建
为便于机器学习的训练,所有样本需要满足特定的样本形式.把将要预测的调控关系基因对按如下形式给出,

其中:TF表示转录因子;TG表示推测可能被TF调控的靶基因.根据特定的训练形式,按以下步骤构建每个基因对的特征向量模型:
(1)提取每个TF及其靶基因TG的表达谱特征向量
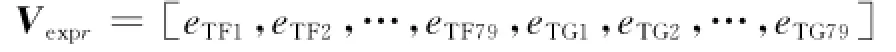
(2)提取由转录因子(氨基酸序列)的20个氨基酸组分构成的特征向量
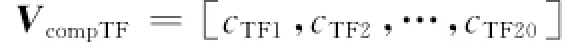
(3)提取由靶基因(DNA序列)的4个碱基组分、16个相邻碱基组分及64个密码子构成的特征向量

将以上3种特征向量组合后的262维向量作为每个基因对的特征向量模型输入分类器中进行分类预测.
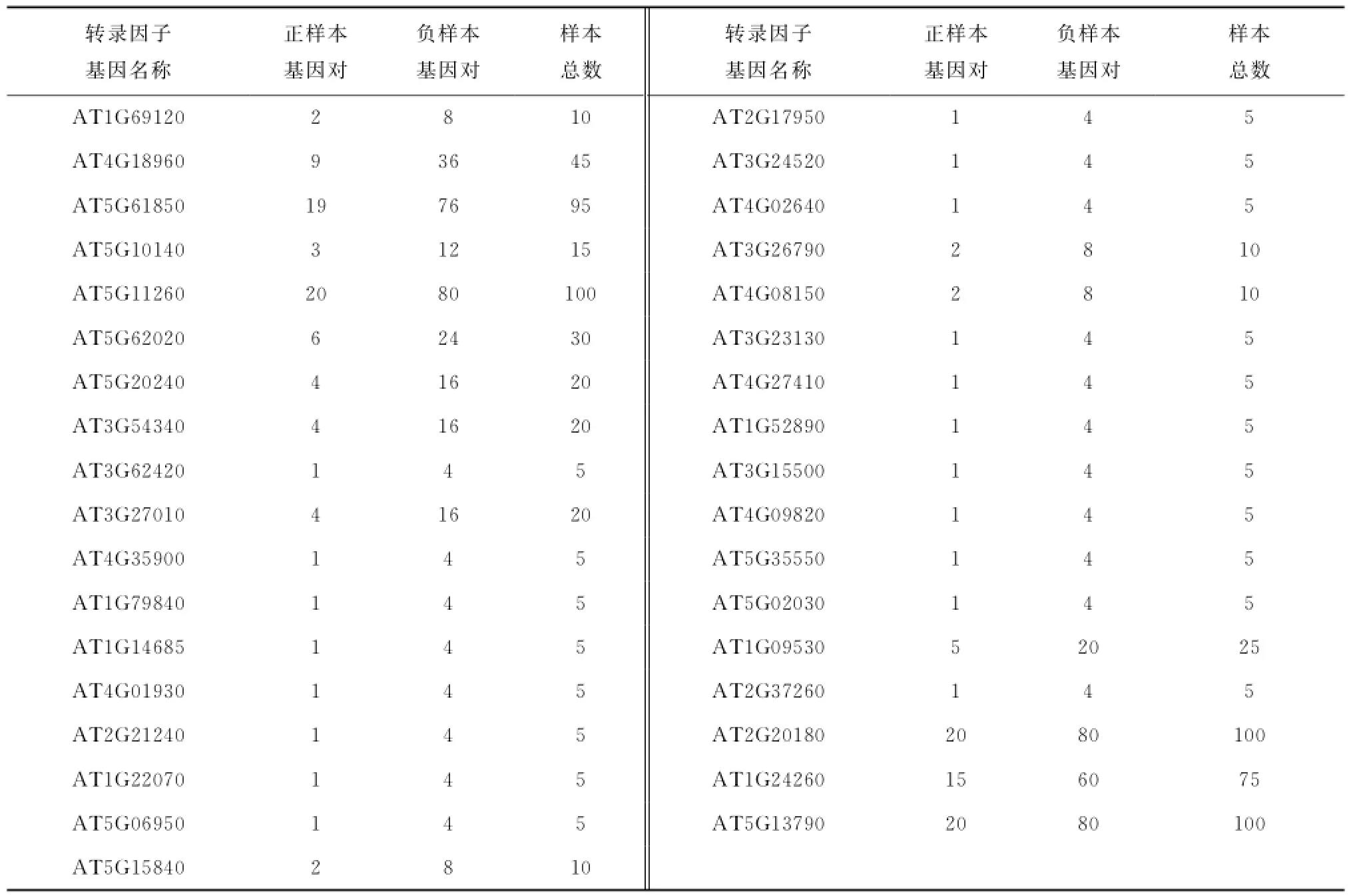
表1 研究中所使用的阳集和阴集数量Tab.1 Number of positive and negative samples collected in study
1.2.3 分类器及检验方法
支持向量机(support vector machine,SVM)是一种具有某些优良特性的“线性分类器”,其数学原理的理论依据是统计学习理论,是一种监督式的机器学习算法.采用SVM软件包[3]运行SVM并进行数据分类和预测工作.径向基函数用作为SVM的核函数,且其可另表示为
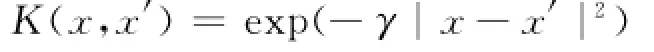
其中,回归参数C以及核宽度参数γ的值是通过在训练集上利用SVM的网络搜索工具不断进行训练而得到的,当训练的预测结果达到最优时,取得此时的两个参数值.
当分类预测工作结束,需要采用一种验证方法来检验和评估所提出的方法在实际应用中的有效性.独立集检验、子样本检验和夹克刀检验是3种最为常用的统计预测方法.其中,夹克刀检验(又称为留一法)通常被认为是最有效的一种方法[4].首先选取一对基因对作为测试样本,剩下的作为训练样本,依次轮流循环,直到所有样本基因对都作为测试样本,此时遍历结束.然后采用敏感度(Sn)、特异性(Sp)以及总准确精度(OA)作为评估预测性能的3个指标,其计算公式分别为:
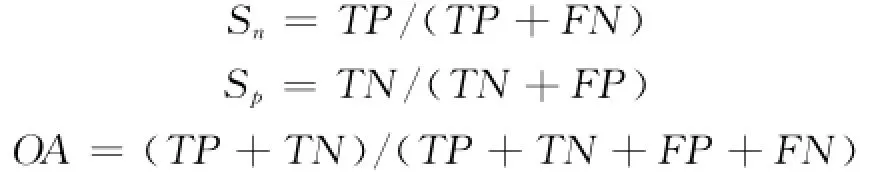
式中,TP、T N、FP、FN分别表示预测正确的阳集样本个数、预测正确的阴集样本个数、预测错误的阳集样本个数以及预测错误的阴集样本个数.
2 结果与讨论
2.1 表达谱的相关性分析
曾有研究者提出一个基本假设:转录因子表达水平的变化将通过转录调控过程影响靶基因表达水平的变化[5].但后来又有研究者提出了另一种观点:微阵列技术是一种高通量的数据分析手段,某种程度上受实验环境等因素影响,并不是非常准确的.转录因子和靶基因在表达谱上的相似性只表示了一种统计依赖性,在某段时间上不一定存在因果关系[6].为观察拟南芥具有调控关系的转录因子和靶基因表达谱数据的相关性情况,随机选取了部分阳集数据并对其进行了相关性分析,如图2所示.图2(a)中具有调控关系的基因表达谱具有一定的相关性,图2(b)中的调控关系转录因子的表达并没有影响靶基因的表达,图2(c)和(d)中只有某一段时间上的表达具有相关性.尽管都是具有调控关系的基因对,但转录因子和靶基因在表达上并不存在直接的因果关系.该现象也表明,仅仅依靠表达谱信息来预测拟南芥的基因调控关系是不充分的.
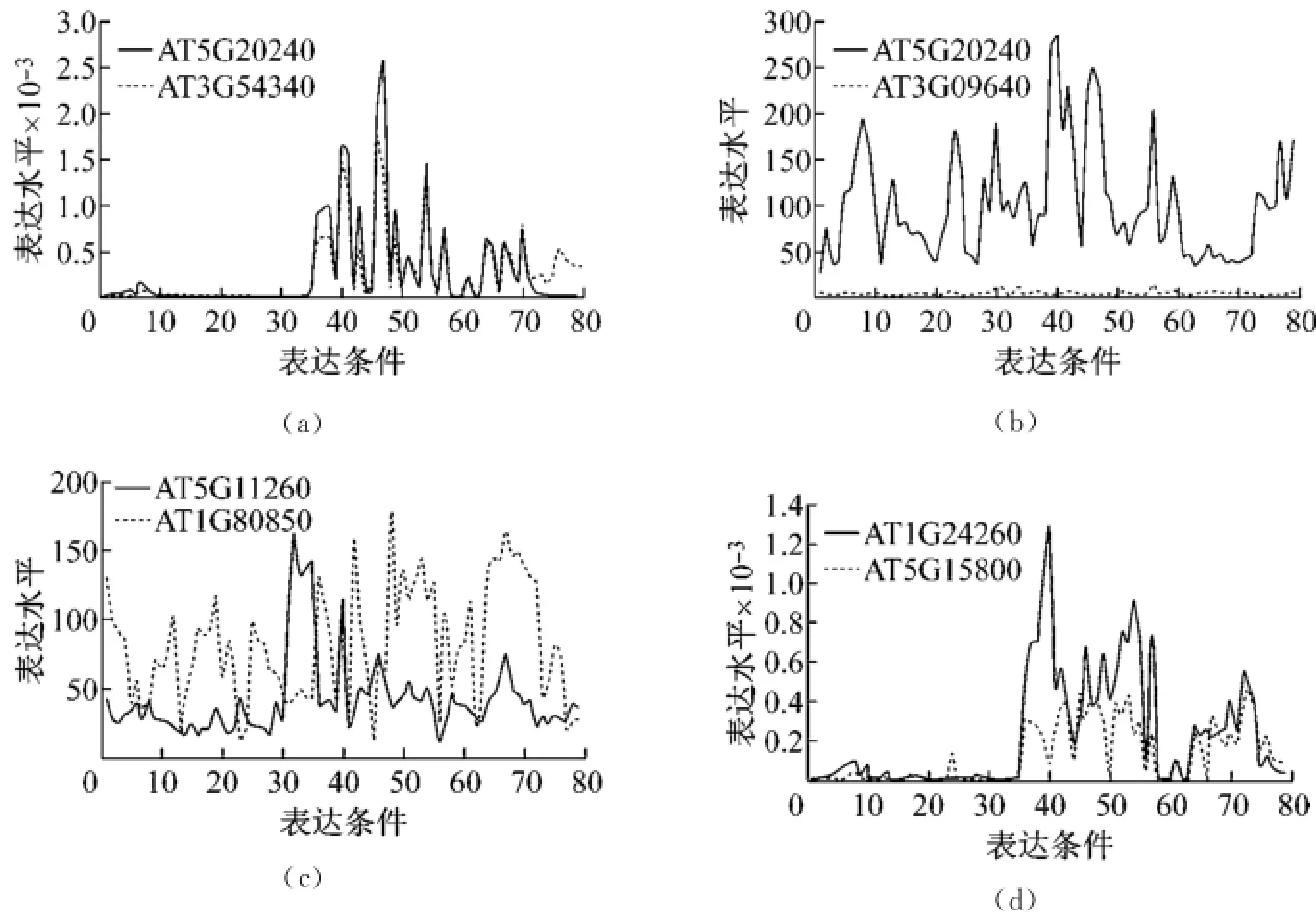
图2 部分阳集样本中转录因子与其靶基因的表达水平Fig.2 The gene expression level of TF and its target for part of the positive samples
2.2 结果分析
所有模型的预测系统都在最优回归参数C=32和核宽度参数γ=3.051 757 812 5E-005时进行.为观察不同特征组合的预测模型对预测结果的影响,计算了不同情况的预测结果,见表2.
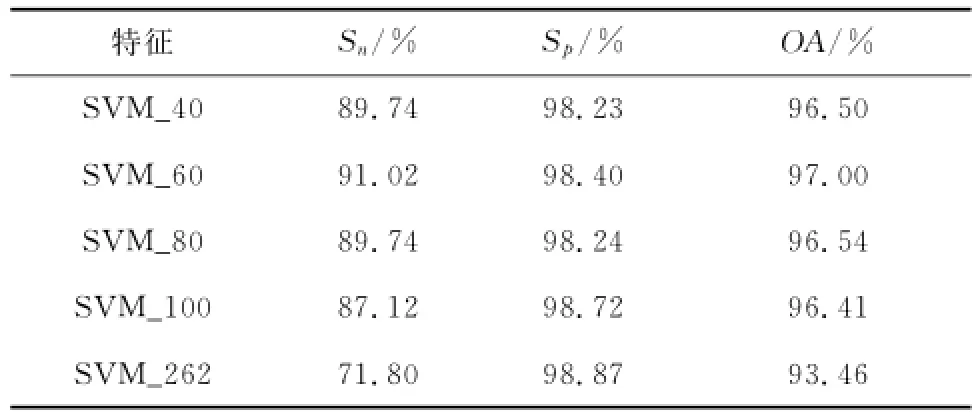
表2 不同特征的预测性能比较Tab.2 Prediction performance comparison using different selected features
在利用机器学习方法对数据进行分类预测中,由于训练样本有限,故若要使机器学习算法具有很好的泛化能力,则样本特征需要尽可能地少[7].利用SVM软件包中的特征选择工具得到每个特征对预测结果的贡献得分,选择了得分最高的60个特征作为最优特征子集,取得的预测准确度为97%,敏感度为91.02%,特异性为98.4%.计算了其他K个特征的预测结果(K=40,80,100,262).由表2可见,各种情况对于特异性的计算结果都比较高,这也是构建特征向量模型时所期待的结果.因为对于拟南芥这种模式植物,已被确定具有调控关系的阳集样本数量有限,而实验室中的实验需耗费大量时间、资金等,所以构建的预测模型应该尽可能地提高特异性的预测结果,才能使预测模型具有更好的可行性.没有进行特征选择的SVM_262的敏感度和总准确度相对较低,这说明选取最优特征子集的方法还是非常有必要的.在选择的60个特征中,包含31个表达数据特征和29个序列特征,这也表明了序列信息特征在拟南芥调控关系的预测中起到了一定的作用.
3 结 语
基于表达谱数据和序列相关信息构建了基因转录调控关系的特征向量模型,通过SVM提出了一种预测拟南芥基因转录调控关系的计算方法.预测结果表明,该方法对预测拟南芥转录调控关系有着良好的表现.目前,由于拟南芥阳集样本的数量有限,故该方法还受到数据的局限性.相比实验方法,计算方法可以节省大量时间和资金,并为实验方法提供一定理论依据.随着生物数据的不断增多,利用计算方法来预测拟南芥基因转录调控关系将会成为实验方法的一个有益补充.参考文献:
[1] Pournara I,Wernisch L.Factor analysis for gene regulatory networks and transcription factor activity profiles[J].BMC Bioinformatics,2007,8:61.
[2] Tan K,Mccue L A,Stormo G D.Making connections between novel transcriptionfactors and their DNA motifs[J].Genome Res,2005,15(2):312-320.
[3] Chang C C,Lin C J.LIBSVM:A library for support vector machines[EB/OL].[2011-08-30].http://www.csie.ntu.edu.tw/~cjlin/libsvm.
[4] Chen C,Chen L,Zou X,Cai P.Prediction of protein secondary structure content by using the concept of Chou’s pseudo amino acid composition and support vector machine[J].Prot Pept Lett,2009,16:27-31.
[5] Liu W L,Li D,Liu Q,et al.A novel parametric approach to mine gene regulatory relationship from microarray datasets[J].BMC Bioinformatics,2010,11(S11):11-15.
[6] Qian J,Dolled-Filhart M,Lin J,et al.Beyond synexpression relationships:Local clustering of time-shifted and inverted gene expression profiles identifies newbiologically relevant interactions[J].J Mol Biol,2001,314(5):1053-1066.
[7] Briesemeister S,Rahnenführer J,Kohlbacher O.Going from where to why-interpretable prediction of protein subcellular localization[J].Bioinformatics,2010,26(9):1232-1238.
(编辑 吕丹)
Study on Computational Methods for Predicting the Regulatory lnteractions Between Transcription Factors and Their Targets in Arabidopsis
YU Xiao-qing
(School of Sciences,Shanghai Institute of Technology,Shanghai 201418,China)
Arabidopsis,an important model plant,which is widely used in the study of plant biology. Based on the gene expression profile and some sequence-based information,a mathematical model was constructed to predict the regulatory interactions in Arabidopsis.Through support vector machine and Jackknife test,the method was proved to have a good performance.This method could provide some theoretical basis for the laboratory study of the regulatory interaction of Arabidopsis.
Arabidopsis;regulatory interactions;support vector machines;computational approach
O 242.1
A
1671-7333(2015)01-0091-04
10.3969/j.issn.1671-7333.2015.01.016
2014-07-14
上海市高校青年教师培育基金资助项目(ZZyyy13017);上海应用技术学院引进人才基金资助项目(YJ2013-32)
于晓庆(1983-),女,讲师,博士,主要研究方向为计算数学,生物信息学.E-mail:xqyu@sit.edu.cn
猜你喜欢
昆明医科大学学报(2022年1期)2022-02-28
中学生数理化·高一版(2021年2期)2021-03-19
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
知识经济·中国直销(2018年8期)2018-08-23
上海农业学报(2017年3期)2017-04-10
数学学习与研究(2017年3期)2017-03-09
中国老区建设(2016年1期)2016-02-28
红领巾·探索(2015年9期)2015-09-10
医学研究杂志(2015年8期)2015-06-22
