
水系锌锂离子液流电池正极材料研究
2015-11-14 00:48赖勤志李先锋张华民
储能科学与技术 2015年5期
赖勤志,李先锋,张华民
(中国科学院大连化学物理研究所,辽宁 大连 116023)
随着社会与经济的发展,人类对能源的需求量显著增加。化石能源的大量消耗,不仅造成了能源资源的匮乏,而且还造成了严重的环境污染。新能源的开发利用,特别是风能、太阳能等可再生能源的研究受到了广泛关注。然而这些可再生能源却受时间和季节的限制,具有不连续、不稳定的特点。大规模可再生能源发电并网将对电网的安全、可靠、高效运行带来极大风险。大规模、长寿命、高效率、低成本的储能技术是实现可再生能源发电普及应用及智能电网建设所急需的核心技术,在各种电化学储能技术的发展历程中,锂离子电池因其优异的性能而具有较好的应用前景[1]。
锂离子电池拥有最高的能量密度,为120~200 W·h/kg[2]。目前锂离子电池多采用锂离子嵌入化合物为电极材料,并以无水且含Li+的有机溶液为电解液。因为Li+能够可逆地在电极活性物质中嵌入或脱出,且不破坏电极材料的晶体结构,其与传统的水系二次电池相比,如铅酸、镍氢电池,表现出良好的循环寿命。另外,无水有机电解液具有更宽的电位窗口和更高的能量。小型锂离子电池已广泛用在如手机、笔记本、数码相机等移动通讯设备上[3-6]。大型锂离子电池被认为最有希望成为电动汽车的动力电池的选择之一。然而,尽管有机系锂离子电池具有诸多优势,但其使用的有机溶剂不仅有毒而且易燃,如果使用不恰当(如过充、过放、短路等),会带来很多安全性问题。此外无水操作环境大大提高了电池制造成本,这些不足均限制了其在大规模储能领域中的应用。针对上述问题,国内外科研工作者把目光转向了水系锂离子电池。
1994年,加拿大的学者提出了一种水溶液可充放锂电池的概念[5-7],他们首先在碱性LiOH水溶液中用嵌锂LiMn2O4制备了Li2Mn2O4,从而打破了以往认为Li2Mn2O4(vs.Li+/Li)的电压为4.0 V,不能在水溶液中稳定存在的传统思路。
水系锂离子电池具有安全性高、成本较低、环境友好等特点[8-10]。此类电池若能研发成功并实用化,将在大规模储能领域中具有良好的应用前景。
锂离子电池正极材料在水中一般都是稳定的。然而,由于质子(即H+)的半径比Li+小,在水溶液中质子可能与Li+同时嵌入到电极材料中。此外,质子的嵌入跟晶体结构及溶液的pH值有很大关系。尖晶石Li1−xMn2O4和橄榄石 Li1−xFePO4不会发生质子共嵌,而脱锂的层状 Li1−xCoO2Li1−xNi1/3Mn1/3Co1/3O2等在pH较低的电解液中深度脱锂的情况下,晶格中会出现一定浓度的质子[11-15]。这个问题可以通过调解溶液的pH值来控制质子嵌入的电位。如Li1−xNi1/3Mn1/3Co1/3O2在pH值大于11的水溶液中是稳定的,Li1−xCoO2在pH大于9的水溶液中是稳定的[7,13]。值得注意的是,在强碱溶液中,LiFePO4会发生分解,但通过碳包覆可以减缓LiFePO4的分解[16-18],碳包覆的橄榄石LiFePO4可以在pH值7~14内使用[7]。
另一方面常用的负极材料有VO2和LiV3O8等,但是通常情况下这些材料的稳定性较差,限制了电池的比容量和循环性能,因此改善负极材料的稳定性是提高水系锂离子电池性能的关键之一。
金属锌具有比能量高、析氢过电位高、循环稳定性好的优势,在电池中得到了广泛的应用。因此,在本文中利用金属锌的沉积溶解反应作为负极电对,正极采用锂离子的嵌入脱嵌,构建锌锂离子液流电池。为了提高体系的能量密度,选择使用碱性体系,在碱性体系中锌的沉积电位为-1.2 V,相比中性溶液更具有优势。本文系统考察了正极LiFePO4材料在碱性电解液中的性能。
1 材料与方法
1.1 电极制备
将活性物质(磷酸铁锂)、导电剂(Super-p)及黏结剂(聚四氟乙烯乳液PTFE)按质量比85︰10︰5混合,加入适量的无水乙醇分散均匀。分散均匀后,于70 ℃水浴加热,待快干时,先用玻璃棒擀压,再用辊压机均匀擀压成膜。截取边长为1 cm的正方形膜,压于洁净的不锈钢网上制成工作电极。在80 ℃烘箱中干燥12 h。
1.2 测试方法
电化学测试在电化学分析仪CHI6O2上进行,研究体系为三电极体系;SEM测试采用JEOL公司生产的型号为Nikon的台式扫描电子显微镜,观察LiFePO4的表面形貌。使用导电碳胶将样品粘附固定在样品台上,通过真空溅射镀金3 min,工作电压10 kV;XRD测试采用丹东浩元仪器有限公司生产的型号为DX-2700的X射线衍射仪。辐射源为Cu靶(Kα1=1.54056 Å,1 Å=0.1nm);管电压40 kV,管电流30 mA,扫描范围:5°~90°,扫描速度:0.02°/s。
2 结果与讨论
2.1 正极电化学动力学参数的研究
电子转移步骤也即电化学反应步骤,是指反应物质在电极/溶液界面得到电子或失去电子,从而还原或氧化成新物质的过程。这一单元步骤包括了化学反应和电荷传递两部分,是整个电极反应过程的核心。因此研究电子转移阻抗规律有重要意义。
在过电势较低的范围内,电流密度与过电势趋近于直线关系,化学反应动力学参数可按照如下所示的关系式计算

式中,Rct为电子转移阻抗,Ω;η为过电位,V;i为电流密度,A/cm2;i0为交换电流密度,A/cm2;R为普通气体常数(8.3145);T为开尔文温度 (293.15 K);n为转移电子数(这里为1);F为法拉第常数(96500 C/mol);Ks为反应速率常数,C0为溶液的本体浓度。
通过测定LiFePO4在不同KOH浓度中的线性扫描曲线来计算反应的电子转移阻抗,研究碱浓度对LiFePO4动力学参数的影响,如图1所示。
根据图1中趋于线性部分求出其线性回归方程,其斜率K的倒数即Rct,再根据关系式(1)~式(3)求出其交换电流密度(i0)及反应速率常数 (Ks)。计算结果见表1。

图1 LiFePO4在不同KOH浓度中的线性扫描曲线Fig.1 Linear sweep voltammograms of LiFePO4 in different concentration of KOH solutions
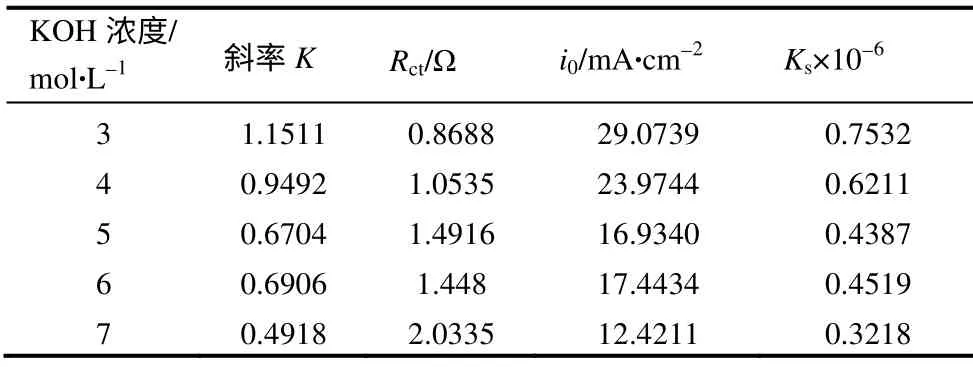
表1 含不同KOH浓度的电解液中的动力学参数Table1 The kinetic parameters of the electrolyte in different concentration of KOH
由表1可以看出,随着KOH 浓度的增大,电子转移阻抗增大,交换电流密度、反应速率常数减小。说明随着KOH浓度的提高,电化学反应越来越难以进行。
2.2 锂离子扩散系数的研究
“化学扩散系数”是一个包含以上扩散过程的宏观的概念,目前被广为使用。化学扩散系数可以由循环伏安法测得,电流与电势的关系符合式(4)。

式中,Ip为峰电流强度,A;n为转移的电子数;A为工作电极面积,cm2;DLi为扩散系数,cm2/s;C0为电解液锂离子浓度,mol/mL;V为循环伏安测试的扫速,V/s。
根据不同扫速下的循环伏安曲线,利用式(4)计算反应的扩散系数。由于在反应过程中,转移的电子数、电解液的浓度都是相同的,所以峰电流强度对扫速的平方根曲线的斜率即反应扩散系数DLi的大小。
分别考察KOH浓度为3~7 mol/L时,在 25 mV/s、50 mV/s、75 mV/s和100 mV/s不同的扫速下进行循环伏安测试,图2(a)~图2(e)分别为溶液3 mol/L、4 mol/L、5 mol/L、6 mol/L、7 mol/L在不同扫速下的循环伏安曲线。图2(f)是峰电流强度与扫速的平方根变化关系曲线,同时将图2(f)中各曲线的斜率值列于表2。


图2 正极在不同扫速下的循环伏安曲线Fig.2 The cyclic voltammograms for positive at different scan rates

表2 图2(f)中各曲线的斜率Table 2 The slope of the curves in Fig.2 (f)
由表2中斜率大小可知,在溶液KOH浓度大于3 mol/L时,锂离子均能快速地在材料内部迁移。
2.3 正极电化学行为的研究
图3为LiFePO4在不同碱浓度条件下的扫速为100 mV/s、电势扫描范围为-0.5~0.7 V的循环伏安曲线。
由图3可知,LiFePO4的氧化峰不明显,原因在于它与析氧电位接近,与析氧峰重合。LiFePO4在不同KOH浓度中的还原峰电位分别为:-0.134 V、-0.104 V、-0.07 V、-0.078 V、-0.067 V,对应的还原峰电流分别为:5.59118 A、6.57524 A、4.71544 A、5.52772 A、5.80105 A。
KOH浓度为3 mol/L、4 mol/L时,因为随着KOH浓度的增大,扩散系数也增大,极化减小,所以还原峰电位正移,反应动力学活性好,峰电流增加;但是当KOH浓度达到一定数值(>4 mol/L),对峰电位值影响不大;从还原峰电流值可以看出,KOH为4 mol/L时LiFePO4的还原峰电流最大,其它浓度对峰电流值影响不大。
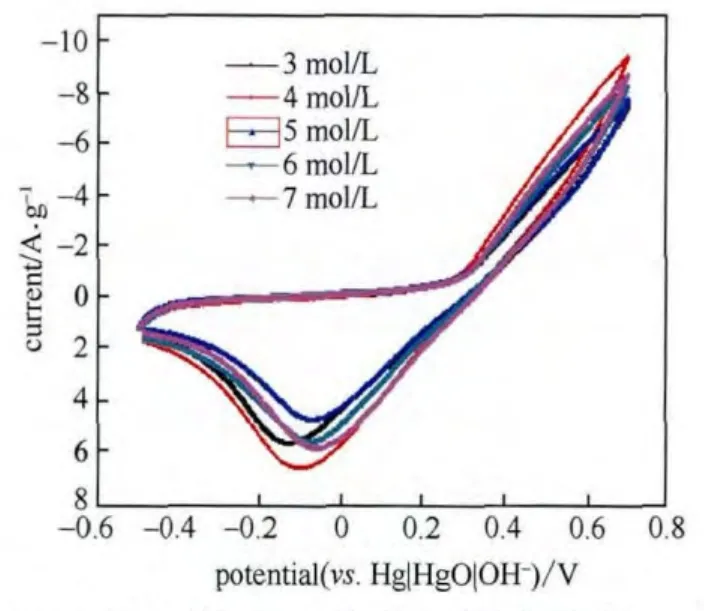
图3 LiFePO4在不同KOH浓度溶液中扫速为100 mV/s的循环伏安曲线Fig.3 The cyclic voltammograms of LiFePO4 in different concentration of KOH, scan rate 100 mV/s
2.4 循环稳定性分析
在对正极材料LiFePO4进行循环伏安测试时,发现LiFePO4在研究体系中衰减很快,稳定性不好。为了研究LiFePO4在不同KOH浓度电解液中的循环稳定性,对其做循环寿命分析,测试了LiFePO4在不同KOH浓度条件下的循环伏安测试,并对比了LiFePO4在不同溶液中的第2圈以及第200圈的循环伏安曲线,如图4所示。
由图4可知,LiFePO4在碱性溶液中,循环稳定性较差,为了探究原因,对正极材料LiFePO4在循环前后的形貌与物性进行了相关的物理表征,SEM测试如图5所示。


图4 LiFePO4在不同KOH浓度溶液中循环稳定性分析Fig.4 The cycle stability of LiFePO4 in different concentration of KOH

图5 循环前(a)和循环后(b)的LiFePO4的扫描电镜照片Fig.5 SEM images of LiFePO4 before (a) and after(b) cycles
通过观察图5(a)、(b)两张图片,可以发现循环后的LiFePO4颗粒粒径明显增大。这说明LiFePO4在含KOH的溶液中,在结构方面发生了变化。
为了进一步研究LiFePO4在含KOH的溶液中在结构等方面到底发生了哪些变化,对循环前后的LiFePO4进行了XRD测试,结果如图6所示。图6中循环前的LiFePO4的XRD图谱为黑色曲线,而红色曲线是循环后的LiFePO4的XRD图谱。
从图6中可以看出,循环后的LiFePO4的衍射峰强度变小是LiFePO4结晶度降低的结果,可能是由于LiFePO4正极材料出现部分锂离子溶出,在磷酸铁锂颗粒中形成了不完整的FePO4,这也是LiFePO4在电解液中衰减比较快的原因。
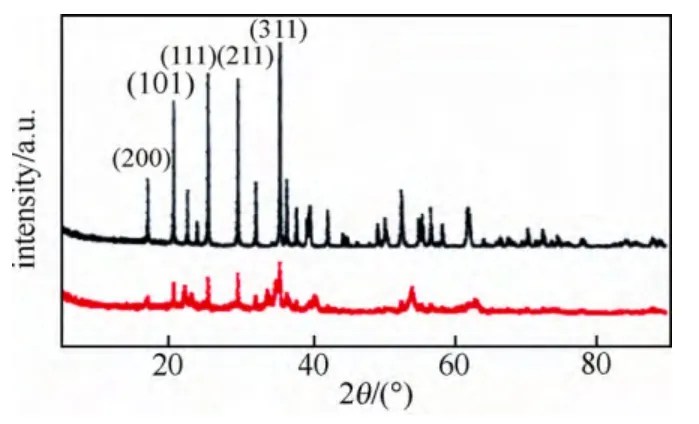
图6 循环前后的LiFePO4正极材料XRD图Fig.6 XRD spectra of LiFePO4 before and after cycles
3 结 论
本工作考察了LiFePO4作为锌锂离子电池正极材料的可行性。结果表明:① 在含不同KOH浓度的电解液中,随着KOH浓度的增加,正极LiFePO4电子转移步骤的电子转移阻抗增大,反应速率常数减小;② 锂离子的扩散系数随着KOH浓度的增加而增大,在KOH浓度较低时扩散系数较小,不利于离子的扩散,KOH浓度较高时扩散系数较大,但当它达到一定浓度,增大KOH浓度对扩散系数的影响变小;③ 正极LiFePO4在碱性溶液中的稳定性较差,原因在于LiFePO4在电解液中发生了溶解,导致电化学性能的逐渐下降。
[1]Liu Weiwei(刘未未),Wang Baofeng(王保峰),Li Lei(李磊).Recent research progress of electrode materials for aqueous lithium-ion battery[J].Energy Storage Science and Technology(储能科学与技术),2014,3(1):7-20.
[2]Wang G J,Zhao N H,Yang L C,et al.Characteristics of an aqueous rechargeable lithium battery (ARLB)[J].Electrochimica Acta,2007,52(15):4911-4915.
[3]Whittingham Stanley M.Lithium batteries and cathode materials[J].Chem.Rev.,2004,104:4271-4301.
[4]Ning L J,Wu Y P,Fang S B,et al.Materials prepared for lithium ion batteries by mechanochemical methods[J].Journal of Power Sources,2004,133(2):229-242.
[5]Tang Zhiyuan(唐致远),Lu Xinghe(卢星河),Zhang Na(张 娜).Doping of LiMn2O4spinel battery materials[J].Chemistry(化学通报),2005,68(5):321-328.
[6]Armand M,Tarascon J M.Building better batteries[J].Nature,2008,451:652-657.
[7]Luo Jiayan(罗加严).水系锂离子电池和电极材料的研究[D].Shanghai:Fudan University,2009.
[8]Wang Gaojun(王高军).Exploration of new rechargeable chemical power source[D].Shanghai:Fudan University,2008.
[9]Chen Shengyao(陈胜尧).水系可充锂离子电池嵌锂化合物的修饰及电化学性能研究[D].Nanjing:Nanjing University Aeronautics and Astronautics,2009.
[10]Wang Shuangcai(王双才),Li Yuankun(李元坤),Liu Shuping(刘述平).Recent research progress on lithium-ion battery positive materials[J].China’s Manganese Industry(中国锰业),2004(3):31-34.
[11]Wu L,Dahn J R,Wainwright D S.Rechargeable lithium batteries with aqueous electrolytes[J].Science,1994,264(5162):1115-1118.
[12]Glanz James.Lithium battery takes to water-and maybe the road[J].Science,1994,264(5162):1084.
[13]Yi Jin(易金),Wang Yonggang(王永刚),Xia Yongyao(夏永姚).An overview of aqueous lithium-ion batteries[J].Chinese Science Bulletin(科学通报),2013,32:3274-3286.
[14]Choi J,Alvarez E,Arunkumar T A,et al.Proton insertion into oxide cathode during chemical delithiation[J].Electrochemical and Solid-State Letters,2006,9(5):A241-A244.
[15]Manthiram A,Choi J.Chemical and structural instabilities of lithium ion battery cathodes[J].Journal of Power Sources,2006,159(1):249-253.
猜你喜欢
传染病信息(2022年6期)2023-01-12
土木工程与管理学报(2021年6期)2022-01-12
中学生数理化(高中版.高二数学)(2020年1期)2020-02-20
山东冶金(2019年5期)2019-11-16
中学生理科应试(2017年2期)2017-04-01
航空材料学报(2017年1期)2017-02-17
肇庆学院学报(2016年5期)2016-03-11
中国资源综合利用(2016年7期)2016-02-03
肿瘤影像学(2015年3期)2015-12-09
中南大学学报(自然科学版)(2012年3期)2012-07-31
