
酶切型稳定同位素标记肽段为内标用于药物代谢酶的绝对定量分析
2015-11-03 07:38刘喜东等
分析化学 2015年10期
刘喜东等
摘 要 利用带有酶切位点的稳定同位素标记肽段作内标,采取3种方式处理样品,考察了不同的样品处理过程对实验结果准确性的影响。首先合成不同长度的带有酶切位点的肽段,考察其酶解过程,实验结果表明,胰蛋白酶的活性位点具有一定的选择性,最终确定目标肽段两端分别加入3个氨基酸所形成的肽段为内标。分别采用在酶解前加入带酶切位点的稳定同位素标记肽段、不带酶切位点的稳定同位素标记肽段以及在酶解过程后、质谱检测前加入不带酶切位点的稳定同位素标记肽段3种方式处理样品。结果表明,采取第一种方式处理样品的测定结果更接近真实值,相对偏差范围更小,可减小蛋白质绝对定量分析的误差,提高分析结果的重现性。
关键词 稳定同位素标记肽段; 绝对定量; 酶切位点; 药物代谢酶
1 引 言
随着蛋白质组学研究的不断深入,以及研究技术的不断创新和发展,定量蛋白质组学研究正逐渐成为该领域方法学研究的热点,使得蛋白质组学的研究内容更加直观和具体[1~3]。鉴于蛋白质分子量较大、结构复杂、种类较多、同源性较高、不易纯化等特点,研究人员多采用生物质谱技术和稳定同位素标记联用技术进行蛋白质的相对定量和绝对定量研究。目前,主要的定量蛋白质组学方法有稳定同位素代谢标记法(Stable isotope labeling with amino acid in cell culture, SILAC)[4,5]、稳定同位素标签标记法(如Isotope coded affinity tags, ICAT; Isobaric tags for relative and absolute quantification, iTRAQ)[6]、以及稳定同位素标记的氨基酸、肽段、蛋白质和肽段串联体(Concatamers of Q peptides, QconCAT)[7,8]作为内标的蛋白质定量方法。
药物代谢酶是催化药物在体内代谢的一类蛋白酶,影响药物在体内的有效性和安全性,在药物开发过程中受到广泛关注[9,10]。近年来,已有文献基于定量蛋白质组学的方法对药物代谢酶进行定量研究[11~15],但是考虑到样品的处理过程对测定结果的影响,不同方法都有各自的缺陷。为了尽量降低样品的处理过程对测定结果的影响,本实验设计带有酶切位点的稳定同位素标记肽段作为内标,使内标肽段和肝微粒体样本经过共同的样品处理过程,使得测定结果更准确可靠。实验中同时对经过不同样品处理过程所得测定结果进行对比分析,探讨样品处理过程对测定结果的影响,提出适用于药物代谢酶及其它蛋白质定量的合理方案。
2 实验部分
2.1 仪器与试剂
Qtrap 5500型串联质谱仪(美国AB SCIEX公司),配有电喷雾离子化源以及Analyst 1.5.2 数据处理软件; Eksigent nanoLC as-2液相系统(美国Eksigent),实验室自制毛细管Trap柱(2 cm×100 μm),Magic TM C18反相色谱柱(10 cm×75 μm,3 μm); MALDI-TOF/TOF 5800质谱仪(美国AB SCIEX公司)。
肽段HEIQRFADLAPIGLPHRVTKDT, IQRFADLAPIGLPHRVTK和RFADLAPIGLPHRV,稳定同位素标记肽段SVFDQDPFLLR*, VHEEIEQVIGR*, FADIVPTNIPHMTSR*, LGRSVFDQDPFLLR*VVK, EAKVHEEIEQVIGR*NRQ和VQRFADIVPTNIPHMTSR*DIK(上海楚肽生物科技有限公司); 甲酸(色谱纯,德国Fluka公司); 尿素、碳酸氢铵、胰蛋白酶、α-氰基-4-羟基肉桂酸(CHCA),色谱纯乙腈(美国Sigma-Aldrich公司); 二硫苏糖醇(美国Promega公司); 碘乙酰胺(美国New Jersey公司)。实验用水为Milli-Q超纯水(美国Millpore公司)。大鼠肝微粒体样品为实验室自制[15]。
2.2 色谱与质谱条件
流动相A相: 乙腈-水-甲酸(2∶98∶0.1, V/V),B相: 乙腈-水-甲酸(98∶2∶0.1, V/V); 流速为380 nL/min。梯度洗脱程序: 0~30 min,95%~70% A; 30~33 min, 70%~20% A; 33~38 min,20% A; 38~40 min,20%~95% A; 40~45 min,95% A。进样体积: 5 μL。
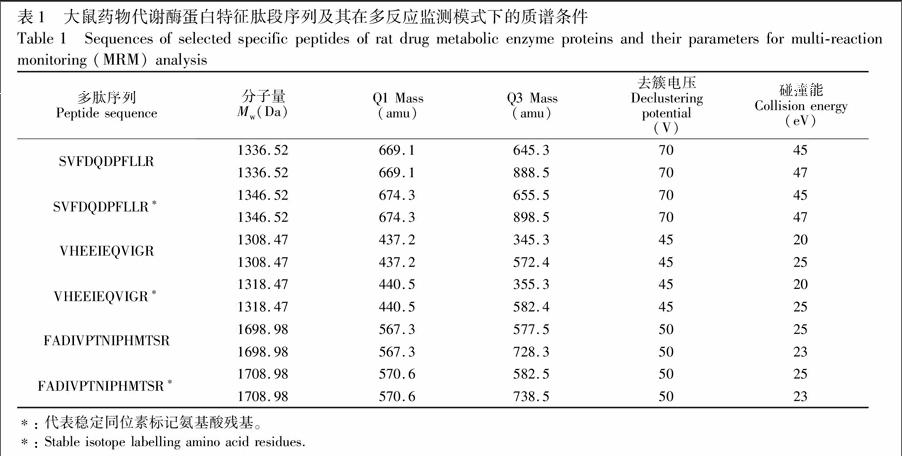
电喷雾离子源温度(TEM): 150℃; 雾化气压力(GS1): 30 psi(0.2 MPa); 气帘气压力(CUR): 15 psi(0.1 MPa); 正离子反应模式; 离子模式喷雾电压(IS): 2200 V; 多反应监测模式离子对、碰撞电压(CE)、去簇电压(DP)等参数见表1。
2.3 特征肽段溶液的配制
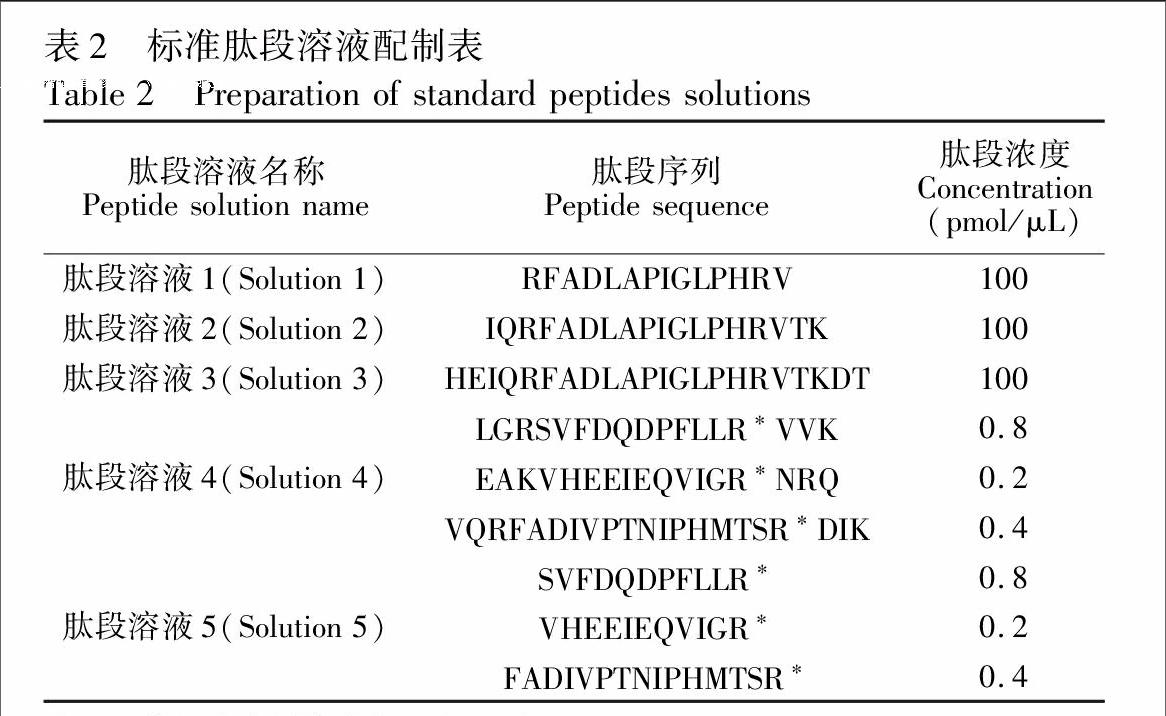
取各肽段标准品,加水溶解,得1 nmol/μL各肽段储备液,于-20℃保存备用。取各肽段储备液,用水稀释,按表2配制相应的肽段溶液。
2.4 考察带有酶切位点肽段的酶解过程
分别取90 μL缓冲溶液(50 mmol/LNH4HCO3)置于3个1.5 mL EP管中,分别加入10 μL肽段溶液1、肽段溶液2和肽段溶液3,再加入2 μL胰蛋白酶溶液(0.5 μg/μL),涡流混匀,在37℃恒温振荡。分别于0, 5, 30和120 min取样10 μL,加入2 μL甲酸酸化,终止反应。以CHCA为基质,进行质谱(MALDI-TOF/TOF)测定。
2.5 3种加入内标肽段形式的方法考察
取3个1.5 mL EP管,编号为A1, A2和A3,每管中加入15 μL大鼠肝微粒体样品(5 mg/mL)、75 μL缓冲溶液Ⅰ(50 mmol/L NH4HCO3、8 mol/L Urea)、8 μL二硫苏糖醇溶液(300 mmol/L); 分别向A1, A2和A3管中分别加入10 μL肽段溶液4、肽段溶液5和水,并涡流混匀,于37℃恒温振荡1 h; 向A1, A2和A3管中分别加入24 μL新鲜配制的碘乙酰胺溶液(300 mmol/L),涡流混匀,于25℃恒温避光振荡40 min; 向A1, A2和A3管中分别加入400 μL缓冲溶液Ⅱ(50 mmol/L NH4HCO3),涡流混匀,再加入胰蛋白酶溶液(0.5 μg/μL),于37℃恒温振荡; 于37℃酶解10 min后,加入甲酸溶液酸化,终止反应; 酶解液通过固相萃取去除杂质,用1 mL 乙腈-水-TFA(80∶ 20∶ 0.1,V/V)溶液洗脱,后于25℃离心热干。向A1、A2管中加入50 μL 0.1%甲酸溶液复溶,向A3管中加入40 μL 0.1%甲酸溶液和10 μL肽段溶液5复溶。取5 μL进样测定。
重复上述步骤,制备酶解0.5, 2, 6和16 h的肝微粒体样品,并进行测定,考察酶解时间对测定结果的影响。
3 结果与讨论
3.1 带有酶切位点肽段的酶解过程考察
在定量蛋白质组学研究中,可采用稳定同位素标记肽段作为内标,实现对目标蛋白的定量分析。在具体实验操作过程中,不同研究者加入内标肽段的步骤并不相同,包括在蛋白样品酶解后、质谱检测前加入内标肽段; 或者在蛋白样品酶解前加入内标肽段,使内标肽段与样品共同进行酶解过程和样品除盐纯化过程。考虑到肽段在样品酶解过程中部分降解,以及样品处理过程中肽段的损失,因此越早将内标肽段和蛋白样品混合,理论测定结果越可靠。由于蛋白质的结构复杂,蛋白质的酶解速率和酶解程度对测定结果也会有影响,稳定同位素标记的蛋白质或肽段串联体等方法即是希望通过同步酶解过程以降低酶解过程带来的误差,但稳定同位素标记的蛋白质或串联体的制备过程相对复杂且成本较高。因此,本研究设计带有酶切位点的稳定同位素标记肽段作为内标,加入到肝微粒体样品中,进行同步酶解和样品处理过程,以最大限度地提高蛋白样品测定的准确性。
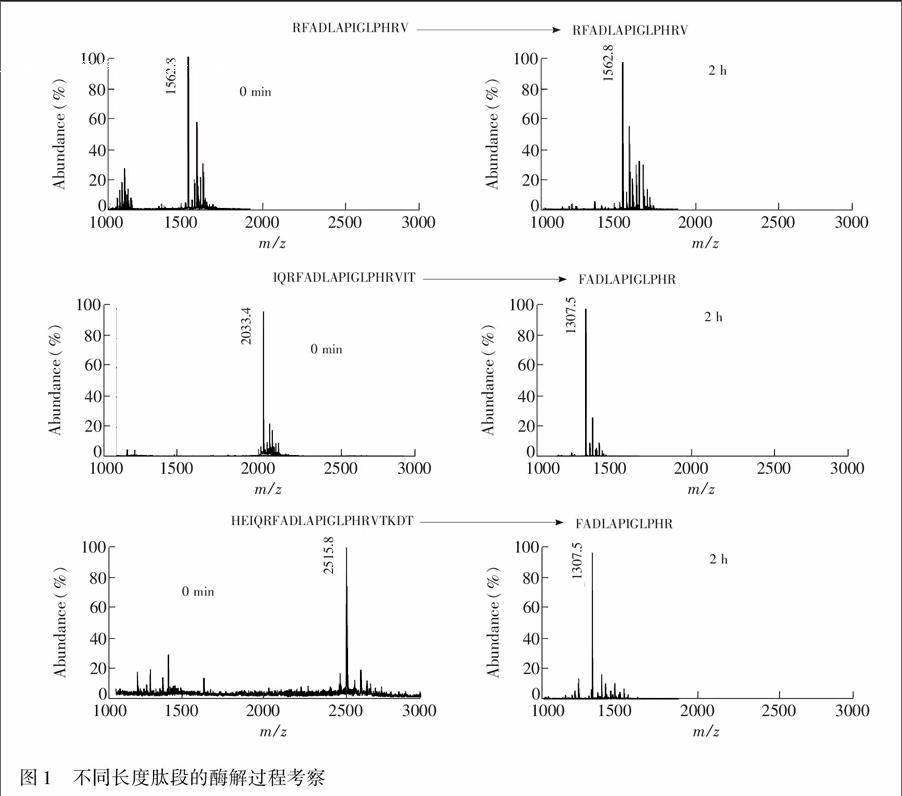
实验中,首先以肽段FADLAPIGLPHR为研究对象,合成具有不同氨基酸数目的带有酶切位点的肽段,考察RFADLAPIGLPHRV、IQRFADLAPIGLPHRVTK和HEIQRFADLAPIGLPHRVTKDT的酶解过程,以选择合适的带有酶切位点的内标肽段。由图1可见,当目标肽段(FADLAPIGLPHR)两端分别添加1个氨基酸时,RFADLAPIGLPHRV不能被酶解产生FADLAPIGLPHR; 当目标肽段两端分别加上3个和5个氨基酸时,IQRFADLAPIGLPHRVTK和HEIQRFADLAPIGLPHRVTKDT都能被酶解产生FADLAPIGLPHR,说明胰蛋白酶的活性位点具有一定的选择性,能选择性的对肽段进行酶切。为了达到内标肽段能被酶切的目的,并考虑到节约实验成本,本实验采用在目标肽段两端分别加上3个氨基酸的研究方案。
实验中考察了肽段IQRFADLAPIGLPHRVTK的酶解过程。如图2所示,肽段IQRFADLAPIGLPHRVTK的两个酶切位点不是同时进行酶切的,这可能与酶切位点处氨基酸的性质有关。通过延长酶解时间,可实现对带有酶切位点肽段的完全酶切。
3.2 药物代谢酶定量方法的建立
实验以UGT1A1、CYP2A1和CYP2D26为研究对象,其水解后可产生的特征肽段分别为SVFDQDPFLLR、VHEEIEQVIGR和FADIVPTNIPHMTSR,通过计算样品中的特征肽段与其已知浓度同位素标记肽段的峰面积比值,得到特征肽段含量,计算得出酶含量。为了提高方法的可靠性,降低仪器因素对测定结果的影响,质谱检测时针对每个肽段选用两个离子对进行定量分析,取两个结果的平均值进行计算。
以3种形式加入同位素标记内标肽段,分别为: 在肝微粒体样品酶解前加入带有酶切位点的稳定同位素标记肽段,使内标肽段和样品进行同步酶解和样品处理过程; 在肝微粒体样品酶解前加入不带有酶切位点的稳定同位素标记肽段,使内标肽段和样品进行同步酶解和样品处理过程; 在肝微粒体样品经过酶解和样品处理后、质谱检测前,加入不带有酶切位点的稳定同位素标记肽段。本研究组曾采用第III种方法对大鼠肝微粒体中细胞色素P450和葡萄糖醛酸转移酶亚型进行了绝对定量分析[15],但没有与前两种方式做系统比较。本实验系统考察了3种形式加入稳定同位素标记肽段做内标对定量结果的影响。
3.3 酶解过程时间长短对测定结果的影响
以上述3种方式加入内标肽段,分别在10 min, 30 min, 2 h, 6 h和16 h终止反应,考察不同酶解作用时间下3种方式加入内标的结果。以特征肽段SVFDQDPFLLR为例,分别计算3种形式加入内标时,各时间点测定结果的平均值,再计算每个时间点的测定值相对于平均值的相对偏差,以时间点为横坐标,各时间点测定结果的相对偏差为纵坐标作图(图3)。图3中各点可表示不同形式加入内标时,样品中特征肽段实际含量的相对变化。
如图3a所示,对于特征肽段SVFDQDPFLLR,随着时间延长,以第Ⅲ种方式加入内标肽段时,测得特征肽段的含量先增加后略降低,在10 min时,蛋白没有完全酶解,测定值负偏差较大,在2 h时蛋白基本完全酶解成肽段,随后产生的特征肽段发生部分降解; 以第Ⅰ、Ⅱ种方式加入内标肽段时,测得特征肽段的含量逐渐增加,说明添加的内标肽段也发生部分降解,特征肽段与内标肽段在样品中变化一致,测定结果更接近真实值。以第Ⅰ和Ⅱ种方式加入内标肽段时测定值基本一致,说明带有酶切位点肽段在样品中迅速酶解。
如图3b所示,对于特征肽段VHEEIEQVIGR,随着时间延长,以第Ⅲ种方式加入内标肽段时,测得特征肽段的含量先增加,后稍许降低,在10 min时,蛋白没有完全酶解,测定值负偏差较大,在2 h时,蛋白基本完全酶解; 以第Ⅱ种方式加入内标肽段时,测得特征肽段的含量逐渐增加,在16 h时正偏差较大,说明蛋白酶解产生的特征肽段降解程度多于内标肽段; 以第Ⅰ种方式加入内标肽段时,测得特征肽段的含量整体趋势逐渐降低,在10 min时,正偏差较大,说明带有酶切位点的内标肽段没有酶解完全,在2 h时,基本酶解。以第Ⅰ、Ⅱ种方式加入内标肽段时,后3个时间点的测定值变化趋势一致,说明带有酶切位点的内标肽段和无酶切位点内标肽段在样品中变化过程一致。
如图3c所示,对于特征肽段FADIVPTNIPHMTSR,随着时间延长,以第Ⅲ种方式加入内标肽段时,测得特征肽段的含量先增加,后显著降低,说明在10 min时,蛋白没有完全酶解,等蛋白完全水解成肽段后,特征肽段又明显降解,使得测定值负偏差较大; 以第Ⅱ种方式加入内标肽段时,测得特征肽段的含量逐渐增加,后基本不变,说明加入的内标肽段也发生降解,降解速度与特征肽段一致; 以第Ⅰ种方式加入内标肽段时,测得特征肽段的含量先增加,后基本不变,说明在10 min时,带有酶切位点的内标肽段没有酶解完全。由于蛋白酶解产生的特征肽段发生降解,当加入带有酶切位点的内标肽段时,特征肽段与内标肽段在样品中变化过程基本一致,测定值更接近真实值。
上述3种特征肽段的定量结果反映了以同位素标记肽段作为内标进行蛋白定量时的普遍问题。如图3a所示,由于蛋白水解产生的特征肽段发生降解,以第Ⅲ种方式加入内标肽段时,测定的结果与理论值相比偏低; 如图3b所示,以第Ⅱ种方式加入内标肽段时,测定的结果与真实值更接近,但是特征肽段从生成后开始稍许降解,而内标肽段从加入后开始稍许降解,测定结果比真实值稍偏大; 如图3c所示,以第Ⅰ种方式加入内标肽段时,由于带有酶切位点的内标肽段需要经过酶切过程,内标肽段与蛋白在样品中的过程更接近一致,使最终的测定结果更接近真实值,相对偏差范围更小。因此,选用带有酶切位点的同位素标记肽段作为内标时,对蛋白质的定量结果更接近真实值,方法的偏差也最小。
3.4 基于定量蛋白质组学的药物代谢酶定量方法总结
利用LC-MS/MS技术,通过计算特征肽段与其同位素标记肽段峰面积的比值来测定蛋白的含量是目前公认的最佳的的蛋白质定量方法,但是需要注意以下问题: (1)需考察特征肽段在样品处理过程中的稳定性,尽量选择在样品长时间处理过程中较稳定的特征肽段; (2)对于内标肽段,可将其与样品混合后再进行样品酶解等处理过程,这样可以降低样品处理过程引入的偏差; (3)内标肽段的纯度可用氨基酸水解法进行标定,保证内标肽段的含量准确; (4)可选择带有酶切位点的标记肽段或标记肽段的串联体(QconCAT)作为内标[16],提高方法的准确性,特别适用于难以确定稳定的特征肽段的蛋白序列,但是需要考察每个内标肽段的酶解过程,这样才能准确评价计算结果的准确性; (5)蛋白样品的处理过程可根据特征肽段的特点进行优化,包括样品的酶解过程的时间和体系中酶浓度等。
References
1 Pennington S R, Wilkins M R, Hochstrasser D F, Dunn M J. Trends Cell Biol., 1997, 7(4): 168-173
2 Wilkins M R, Sanchez J C, Gooley A A, Appel R D, Humphery-Smith I, Hochstrasser D F, Williams K L. Biotechnol. Genet. Eng. Rev., 1996, 13: 19-50
3 WEI Jun-Ying, ZHANG Yang-Jun, ZHAO Yan, CHEN Xi-Shu, MA Yan, YING Wan-Tao, QIAN Xiao-Hong. Chinese J. Anal. Chem., 2012, 40(1): 59-65
卫军营, 张养军, 赵 炎, 陈希曙, 马 岩, 应万涛, 钱小红. 分析化学, 2012, 40(1): 59-65
4 ZHU Jin-Lei, ZHANG Kai, HE Xi-Wen, ZHANG Yu-Kui. Chinese J. Anal. Chem., 2010, 38(3): 434-441
朱金蕾, 张 锴, 何锡文, 张玉奎. 分析化学, 2010, 38(3): 434-441
5 ZHOU Yuan, SHAN Yi-Chu, ZHANG Li-Hua, ZHANG Yu-Kui. Chinese Journal of Chromatography, 2013, 31(6): 496-502
周 愿, 单亦初, 张丽华, 张玉奎. 色谱, 2013, 31(6): 496-502
6 CAO Dong, ZHANG Yang-Jun, QIAN Xiao-Hong. Journal of Chinese Mass Spectrometry Society, 2008, 29(3): 185-191
曹 冬, 张养军, 钱小红. 质谱学报, 2008, 29(3): 185-191
7 Rivers J, Simpson D M, Robertson D H, Gaskell S J, Beynon R J. Mol. Cell Proteomics, 2007, 6(8): 1416-1427
8 Austin R J, Chang D K, Holstein C A, Lee L W, Risler J, Wang J H, Adams L, Krusberski N B, Martin D B. Proteomics, 2012, 12(13): 2078-2083
9 Emoto C, Murase S, Iwasaki K. Xenobiotica, 2006, 36(8): 671-683
10 Kanayama N, Kanari C, Masuda Y, Ohmori S, Ooie T. Xenobiotica, 2007, 37(2): 139-154
11 Ardjomand-Woelkart K, Kollroser M, Li L, Derendorf H, Butterweck V, Bauer R. Anal. Bioanal. Chem., 2011, 400(8): 2371-2381
12 Groer C, Busch D, Patrzyk M, Beyer K, Busemann A, Heidecke C D, Drozdzik M, Siegmund W, Oswald S. J. Pharm. Biomed. Anal., 2014, 100: 393-401
13 Al Ali A, Touboul D, Le Caer J P, Schmitz-Afonso I, Flinois J P, Marchetti C, De Waziers I, Brunelle A, Laprevote O, Beaune P. Anal. Bioanal. Chem., 2014, 406(20): 4861-4874
14 Liu X D, Hu L H, Ge G B, Yang B, Ning J, Sun S X, Yang L, Pors K, Gu J K. Proteomics, 2014, 14(16): 1943-1951
15 LIU Xi-Dong, ZHU Jun, CONG Yu-Ting, HU Liang-Hai, YE Ming-Liang, GU Jing-Kai, ZOU Han-Fa. Chinese J. Anal. Chem., 2014, 42(1): 10-15
刘喜东, 朱 俊, 丛宇婷, 胡良海, 叶明亮, 顾景凯, 邹汉法. 分析化学, 2014, 42(1): 10-15
16 Wei J Y, Ding C, Zhang J, Mi W, Zhao Y, Liu M W, Fu T Y, Zhang Y J, Ying W T, Cai Y, Qin J, Qian X H. Anal. Bioanal. Chem., 2014, 406(17): 4183-4193
