
自身免疫多腺体综合征1例
2015-10-31 06:20姜祎群孙建方
中国麻风皮肤病杂志 2015年10期
刘 白 吴 琼 张 韡 姜祎群孙建方
自身免疫多腺体综合征1例
刘 白 吴 琼 张 韡 姜祎群∗孙建方∗
患者,女,24岁。皮肤黑变2年余。皮肤科检查:全身弥漫性色素沉着,腋下、腹股沟等褶皱部位及掌纹、关节处明显,口唇及齿龈黏膜变黑,甲黑线,阴毛、腋毛少,第二性征有缺陷。甲状腺球蛋白降低,抗甲状腺过氧化物酶抗体升高;皮质醇水平下降;促肾上腺皮质激素水平升高;甲状腺B超示桥本甲状腺炎。诊断为自身免疫多腺体综合征,给予泼尼松口服治疗,症状好转。
自身免疫多腺体综合征; 色素沉着; APS-II型
自身免疫多腺体综合征(autoimmune polyglandular sydrome,APS)系指同一个体有2个或者2个以上内分泌腺体在自身免疫性炎症侵袭下先后或同时发生功能减退,可累及甲状腺、甲状旁腺、肾上腺皮质、性腺、胰腺β细胞、胃壁细胞等。1,2现报道累及甲状腺和肾上腺的自身免疫多腺体综合征1例。
1 临床资料
患者,女,24岁。因皮肤黑变2年余就诊。2年前患者无明显诱因出现臀部皮肤色素沉着,未予处理,色素沉着面积逐渐扩大,1年后全身弥漫性色素沉着,关节处及褶皱部位色素沉着明显,口唇及齿龈黏膜变黑。自述无不适,否认系统性疾病史。进一步问诊和体检,发现患者精神抑郁,表情淡漠,体毛少,第二性征有缺陷。门诊考虑诊断“Addison病”,并进一步行相应实验室检查。
体格检查:生命体征平稳,神志清楚,表情淡漠,查体合作。甲状腺Ⅰ度肿大,质硬。心肺腹检查未及明显异常,月经正常。皮肤科检查:全身弥漫性色素沉着,掌纹、关节处以及腋下、腹股沟等褶皱部位明显,口唇及齿龈黏膜变黑,20甲甲板色深,甲黑线,阴毛、腋毛少,第二性征有缺陷(图1)。
实验室检查:甲状腺功能结果示游离三碘甲状腺原氨酸(FT3)5.64 pmol/L(3.10~6.80 pmol/L),游离甲状腺素(FT4)13.29 pmol/L(12.00~22.00 pmol/ L),促甲状腺激素(TSH)6.450 m IU/L(0.270~4.200 m IU/L),甲状腺球蛋白(TG)0.10 ng/m L(1.4~78.0 ng/mL),抗甲状腺过氧化物酶抗体(anti-TPO)>600 IU/mL(<34.0 IU/mL),促甲状腺素受体抗体(TRAb)0.1 IU/L(0~1.5 IU/L);连续2天查晨8点皮质醇(Cortisol)结果分别为124.00 nmol/L、6 nmol/L(170.0~440.0 nmol/L);两次促肾上腺皮质激素(ACTH)检查结果分别为1171.0 pg/mL、19.03 pg/mL(7.2~63.3 pg/mL);甲状腺B超:双侧甲状腺包膜完整,内部回声增粗,不均匀成网络样改变,血流信号较丰富;双侧甲状腺弥漫性病变,考虑桥本甲状腺炎。肝肾功、抗核抗体谱、免疫球蛋白及补体未见异常,性激素水平正常;肾上腺CT示双侧肾上腺未见明显异常。组织学表现表皮基底层色素增加,未见真皮内色素沉积(图2)。
2 诊断及治疗
结合患者临床表现、相关实验室检查及内分泌科会诊意见,诊断为“自身免疫多腺体综合征”。予泼尼松5 mg日2次,优甲乐150μg日1次口服治疗,2周后症状好转,全身色素沉着减退,口腔及齿龈黏膜接近正常颜色。复查时发现血钾高达7.1 mmol/L,遂住院治疗。住院期间,予氢化可的松静脉点滴及降血钾治疗。血钾降至正常后,继续予泼尼松5 mg日2次口服治疗,血钾再次升高至6.5 mmol/L,后改为等量氢化可的松口服,血钾未再升高。激素、优甲乐定期减量,现维持氢化可的松早10 mg,下午5 mg,优甲乐50μg/d口服。患者情绪逐渐好转,全身皮肤外观接近正常,褶皱部位及齿龈黏膜色素沉着明显减轻,甲黑线显著改善。
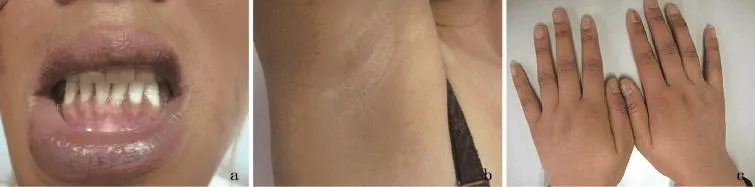
图1 a:口唇及齿龈黏膜变黑;b:腋下褶皱部位色素加深,腋毛少;c:手背皮肤色深,指关节背面尤其明显,甲板颜色加深,甲黑线
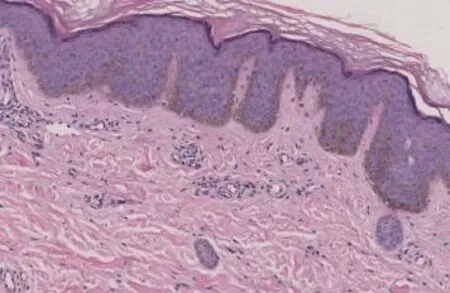
图2 表皮基底层色素增加,未见真皮内色素沉积(HE,×100)
3 讨论
根据患者的起病年龄、临床特征及遗传特性,APS可分为I~IV型。3APS-I型又称自身免疫性多内分泌腺体病合并念珠菌病及外胚层营养不良,2,4临床定义为至少出现以下标准三联征中的两种疾病:慢性皮肤黏膜念珠菌感染(持续3个月以上)、甲状旁腺功能减退和原发性肾上腺皮质功能减退(Addison病)。其他合并疾病包括性腺发育不良、非内分泌系统自生免疫性疾病(干燥综合征、青少年型类风湿关节炎)等。此型多于儿童时发病,女性多见,属常染色体隐性遗传。APS-II型又称Schmidt综合征,可于任何年龄段发病,高峰在20~30岁,女性发病率高于男性。APS-III型是自身免疫性甲状腺疾病(autoimmune thyroid diseases,AITD)并发其他自身免疫性疾病,5,6如1型糖尿病(T1MD)、恶性贫血、白癜风、秃发、系统性自身免疫性疾病。其特点是内分泌和非内分泌器官特异性自身免疫失调,T细胞浸润导致的靶细胞功能障碍,可由环境因素诱发,是不完全外显的常染色体显性遗传,分为4个亚型。APS-IV型最为少见,是Addison病合并前三型之外的自身免疫性疾病,7而无念珠菌感染、AITD或者T1MD。
结合该患者的临床特点,患者青年女性,精神抑郁,体检发现表情淡漠,甲状腺Ⅰ度肿大,质硬,TSH、 anti-TPO水平升高,TG低于正常,甲状腺B超提示双侧甲状腺弥漫性病变,桥本甲状腺炎和甲状腺功能减退诊断明确;有明显的皮肤黏膜色素沉着,两次查血浆皮质醇均低于正常,呈高ACTH血症,“Addison病”诊断明确。结合患者临床表现及实验室检查,存在上述甲状腺、肾上腺2个腺体的自身免疫性疾病,故符合APS-II型的诊断。APS-II型指出Addison病和自身免疫性甲状腺疾病(AITD),还可以出现T1MD、性腺功能衰竭、恶性贫血、重症肌无力及卵巢功能衰竭,其中AITD包括Graves病(GD)、桥本甲状腺炎(HT)、无症状自身免疫性甲状腺炎、特发性黏液水肿和甲状腺相关眼病(TAO)。2,8AITD的特点为出现甲状腺自身抗体和淋巴细胞浸润甲状腺组织,其发病与环境和遗传因素密切相关,其遗传易感性与HLA复合体等位基因关系密切,常与HLA-DR3或DR4单倍体有关,受多基因位点影响,属多基因遗传,为不完全外显常染色体显性遗传。
APS-II型为多基因遗传,发病机制尚不明确,自身免疫敏感个体暴露于环境或者自身的触发条件后,器官特异性自身抗体出现。8目前尚无法根治,只能早期明确诊断,治疗相关疾病、改善症状、缓解病人痛苦,并应长期随访,关注其他腺体是否发生自身免疫病变,早发现,早治疗。1,2本病少见,由于皮肤黑变容易受到患者关注而求治于皮肤科,因此值得皮肤科医生学习和关注。
1 Cutolo M.Autoimmune polyendocrine syndromes.Autoimmun Rev,2014,13(2):85-89.
2 Lebovitz HE.Autoimmune polyglandular syndromes:interplay between the immune and the endocrine systems leading to a diverse set of clinical diseases and new insights into immune regulation.Diabetes Technol Ther,2013,15(Suppl 2):S2-21-S2-28.
3 Dittmar M,Kahaly GJ.Polyglandular autoimmune syndromes:immunogenetics and long-term follow-up.J Clin Endocrinol Metab,2003,88(7):2983-2992.
4Weiler FG,Dias-da-Silva MR,Lazaretti-Castro M.Autoimmune polyendocrine syndrome type 1:case report and review of literature.Arq Bras Endocrinol Metab,2012,56(1):54-66.
5 Kim SJ,Kim SY,Kim HB,et al.Polyglandular Autoimmune Syndrome Type IIIwith Primary Hypoparathyroidism.Endocrinol Metab(Seoul),2013,28(3):236-240.
6 Kahara T,Wakakuri H,Takatsuji J,et al.Autoimmune polyglandular syndrome type 3 with anorexia.Case Rep Endocrinol,2012,2012:657156.
7 Rojas J,Villalobos M,Martinez MS,et al.Successful management of insulin allergy and autoimmune polyendocrine syndrome type 4 with desensitization therapy and glucocorticoid treatment:a case report and review of the literature.Case Reports Immunol,2014,2014:394754.
8Banzal S,Singhai A.Shock:A possible presentingmanifestation of autoimmune polyendocrine syndrome type II.Indian J Crit Care Med,2014,18(5):326-327.
(收稿:2015-04-23 修回:2015-07-02)
·临床研究·
Autoimm une polyglandular syndrome:a case report
LIU Bai,WU Qiong,ZHANGWei,et al.Institute of Dermatology,Chinese Academy ofMedical Sciences and Peking Union Medical College,Nanjing,210042
A 24-year-old woman presented with hyperpigmentation of skin formore than 2 years.Physical examination revealed diffuse hyperpigmentation throughout the whole body,especially the axillar and inguinal folds,skin over joints,and palmar creases.Oralmucosa and gingivae was involved,along with longitudinalmelanonychia.Pubic hair and armpithairwere sparse,which indicated defectof secondary sexual characteristics.Auxiliary examination showed decreased thyroglobulin and increased anti-thyroid peroxidase antibody and ACTH.B ultrasonic showed Hashimoto's thyroiditis.The diagnosis of autoimmune polyglandular syndrome wasmade.After oral prednisone,symptoms improved greatly.
autoimmune polyglandular syndrome;hyperpigmentation;type of APS-II
中国医学科学院北京协和医学院皮肤病研究所,南京,210042
猜你喜欢
现代临床医学(2022年1期)2022-02-12
波谱学杂志(2021年3期)2021-09-07
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年4期)2021-07-23
保健与生活(2021年5期)2021-04-12
天津医科大学学报(2021年2期)2021-03-29
昆明医科大学学报(2020年12期)2021-01-26
中华养生保健(2020年9期)2021-01-18
儿童故事画报·智力大王(2020年3期)2020-04-26
中国医科大学学报(2020年4期)2020-01-08
