
酿酒酵母APA1的定点突变以及与增强型绿色荧光蛋白EGFP基因的共表达
2015-10-29 02:47姚明月刘延琳
食品科学 2015年23期
马 涛,姚明月,刘延琳,2,*
(1.西北农林科技大学葡萄酒学院,陕西 杨凌 712100;2.陕西省葡萄与葡萄酒工程技术研究中心,陕西 杨凌 712100)
酿酒酵母APA1的定点突变以及与增强型绿色荧光蛋白EGFP基因的共表达
马涛1,姚明月1,刘延琳1,2,*
(1.西北农林科技大学葡萄酒学院,陕西 杨凌712100;2.陕西省葡萄与葡萄酒工程技术研究中心,陕西 杨凌712100)
应用基因突变技术向酿酒酵母APA1基因中引入同义突变,为进一步研究APA1表达量与酿酒酵母硫化氢产量的关系提供实验基础。从酿酒酵母S288c中利用聚合酶链式反应扩增APA1基因,与EGFP基因融合构建表达载体。以该载体为模板,通过一步法点突变技术向APA1基因中分别引入APA1-1/2/3/4 4 个突变。测序结果表明突变位点与预期结果一致。成功获得了APA1的4 个同义突变。一步法点突变技术是一种高效的定点突变方法。分别转化酿酒酵母YS59,荧光显微镜下可见绿色激发荧光,表明APA1-1/2/3/4与EGFP基因共表达。
载体构建;酿酒酵母;同义突变
APA1基因编码ADP硫酸化酶,催化进入酵母细胞的SO42-生成腺苷-5'-磷硫酸盐(adenosine 5'-phosphosulfate,APS),最终经过其他酶催化生成H2S[1]。H2S在葡萄酒中的存在被认为是一种感官上的缺陷。H2S的产生与硫代谢相关。硫代谢参与正常酵母细胞代谢活动,通过将无机硫转化成有机硫满足酵母细胞对含硫氨基酸的需求,并完成后续正常生命代谢活动。这一正常代谢伴随着中间产物S2-的产生,即H2S的形成。H2S给葡萄酒带来臭鸡蛋、大蒜、洋葱等不良气味,且味觉阈值低,影响消费者的感官品尝[2-3]。
传统去除葡萄酒中H2S的方法主要是铜沉淀和惰性气体剥离的方法,但是经铜沉淀处理的葡萄酒需后期分离且铜离子浓度受到法定限制,惰性气体剥离没有特异性,影响葡萄酒正常香气,给这些技术的应用带来了限制[4]。因此研究H2S分子机制,认识其产生机理就有极大的实践意义[5-6]。
基因定点突变技术是在基因预定的位点,进行精确地替换、插入或缺失一定长度的核苷酸片段,去研究蛋白质结构与功能的关系。而同义突变不影响蛋白质的序列组成,被认为不影响蛋白质的功能。但是近年来许多文章报道同义突变影响蛋白表达量[7-11]。Bentele等[12]以大肠杆菌为材料,在起始密码子后18 个碱基序列内引入同义突变,并将该基因与黄色荧光蛋白连接,构建载体表达后发现不同同义突变的表达量相差达60 倍之多。为了研究蛋白翻译效率,Qian Wenfeng等[9]将黄色荧光蛋白基因插入酵母染色体上,然后分别用表达载体转化,这些载体上携带发生了同义突变的红色荧光蛋白基因,发现由于引入的同义突变不同,造成了黄色荧光蛋白表达量的不同。本研究通过一步法点突变技术[13-14],向APA1基因中引入同义突变,并将其与增强型绿色荧光蛋白(enhanced green fluorescence protein,EGFP)融合,通过绿色荧光蛋白的不同强度得到表达量不同的APA1基因,为进一步研究该基因表达量与H2S产量的关系提供参考。
1 材料与方法
1.1菌种
大肠杆菌(Escherichaa coli)DH5α生工生物工程(上海)股份有限责任公司;酿酒酵母YS59(MATa、ura3、trp1、leu2、his4)、酿酒酵母标准菌株S288c、质粒pY16(图谱见图1)西北农林科技大学葡萄酒学院微生物实验室保藏;质粒pGEFP-N1、pMD19日本TaKaRa公司。
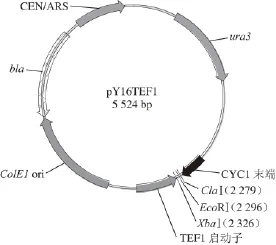
图1 pY16质粒图谱Fig.1 Plasmid profile of pY16
1.2试剂与仪器
HSTM Taq、限制性内切酶(XbaⅠ、ClaⅠ、EcoRⅠ、DpnⅠ)、T4 DNA连接酶、SD-Ura培养基日本TaKaRa公司;聚合酶链式反应(polymerase chain reaction,PCR)产物纯化试剂盒天根生化科技(北京)有限公司;S. c. EasyComp Transformation Kit美国Invitrogen公司;其他试剂均为国产分析纯。
PCR扩增仪美国Bio-Rad公司;荧光显微镜Leica公司;电泳仪北京六一仪器厂;凝胶成像系统美国Thermo公司。
1.3方法
1.3.1APA1基因克隆
1.3.1.1酵母菌的培养以及DNA提取
用YPD固体培养基划线培养标准菌株S288c,挑取单克隆,YPD液体培养2 d后,室温、12 000×g离心2 min收集菌体,提取DNA[15-16]。
1.3.1.2引物设计
根据美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)数据库中S288c菌株的全基因组数据,用Primer 5.0设计引物扩增APA1基因,并引入酶切位点XbaⅠ和EcoRⅠ。上游引物F1:5'-GCTCTA GAATGAGTATCCCCGCTGAC-3',下游引物R1:5'-CG GAATTCATAGTTGTATTCGTTTGG-3'。
1.3.1.3APA1基因扩增
用提取的S288c的基因组DNA为模板,扩增APA1基因。扩增条件:预变性94 ℃ 3 min;变性94 ℃ 30 s,退火55 ℃ 30 s,延伸72 ℃ 1 min。琼脂糖凝胶电泳后,割胶回收纯化目的条带。
1.3.1.4克隆载体的连接、转化、鉴定
将回收产物加A尾后与pMD19载体连接,转化大肠杆菌DH5α,接种到含有Amp的抗性平板上筛选。12 h后挑取单克隆进行菌落PCR鉴定,挑取阳性克隆于含有Amp抗性的LB液体培养基37 ℃、160 r/min培养。12 h后离心收集菌体,提取质粒并送测序。命名为T-pMD19-APA1。
1.3.2EGFP基因扩增
1.3.2.1引物设计和EGFP基因扩增
根据NCBI数据库中的信息,用Primer5.0设计引物扩增EGFP基因,并引入酶切位点EcoRⅠ和ClaⅠ。上游引物F2:5'-GCGAATTCGGTGGTGGTATGGTGAGCAAGGGC-3';下游引物R2:5'-CCATCGATTTACTTGTACAGCTCGTC-3'。以质粒pGEFP-N1为模板,扩增EGFP基因。扩增条件同1.3.1.3节。
1.3.2.2克隆载体的连接、转化、鉴定
方法同1.3.1.4节,载体命名为T-pMD19-EGFP。
1.3.3表达载体pY16TEF1-EGFP-APA1的构建
用XbaⅠ和EcoRⅠ、EcoRⅠ和ClaⅠ以及XbaⅠ和ClaⅠ分别酶切载体T-pMD19-APA1、T-pMD19-EGFP以及pY16,琼脂糖凝胶电泳回收目的酶切片段,T4连接酶连接过夜后转化大肠杆菌DH5α,接种到含有Amp的抗性平板上筛选。12 h后挑取单克隆进行菌落PCR鉴定,挑取阳性克隆于含有Amp抗性的LB液体培养基37 ℃、160 r/min培养。12 h后离心收集菌体,提取质粒酶切鉴定[17-18],命名为pY16TEF1-EGFP-APA1。
1.3.4同义突变
以pY16TEF1-EGFP-APA1为出发载体,通过一步法,设计不同引物,引入突变位点,扩增载体,得到带缺口的定点突变产物APA1-1/2/3/4,用DpnⅠ处理产物,然后纯化后,转化DH5α测序[19-20]。同义突变位点以及引物信息如表1所示。
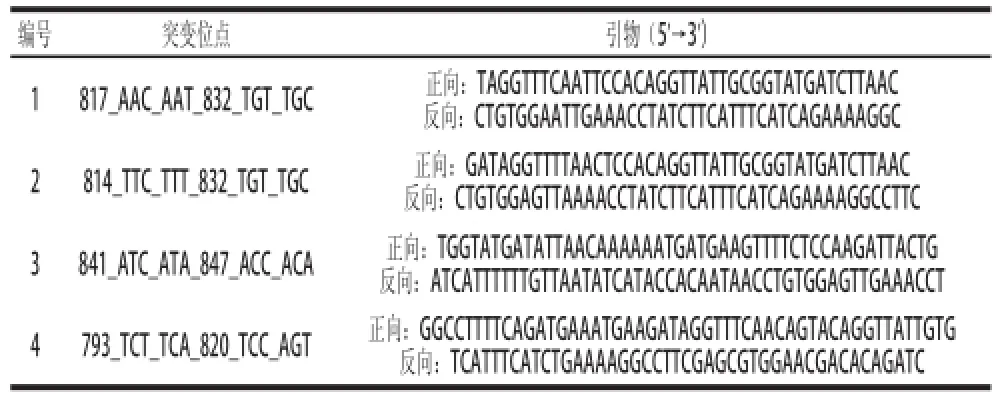
表1 同义突变位点信息Table 1 Information about synonymous mutation sites
1.3.5pY16TEF1-EGFP-APA1转化酵母细胞
提取pY16TEF1-EGFP-APA1-1/2/3/4,用S. c. EasyComp Transformation Kit完整细胞转化法[21],转化酿酒酵母YS59,通过SD-Ura培养基,筛选阳性转化子。详细方法参见S. c. EasyComp Transformation Kit。
1.3.6APA1基因表达鉴定
用YPD液体培养基培养YS59转化子,离心收集菌体,用磷酸盐缓冲液(phosphate buffered saline,PBS)悬浮菌体,荧光显微镜下观察[22]。
2 结果与分析
2.1APA1基因和EGFP基因的克隆
以提取的酿酒酵母S288c基因组为模板,用引物F1和R1扩增得到预期目的片段,大小约为1 000 bp左右。以质粒pGEFP-N1为模板,用引物F2和R2扩增得到预期基因片段,大小约为750 bp左右。扩增产物大小均符合预期,如图2所示。

图2 2 APA1APA1和EGFPEGFP基因的PCR扩增PCRFig.2 PCR amplification of APA1 and EGFP
将上述基因扩增片段分别与pMD19-T载体连接,转化大肠杆菌DH5α,菌落PCR验证后,提取质粒,经XbaⅠ和EcoRⅠ酶切后,得到预期载体片段和APA1基因片段。用EcoRⅠ和ClaⅠ酶切后,得到预期EGFP基因片段,如图3所示。将测序结果与GenBank(登录号:NM_001178695)序列进行比对,相似度100%,表明APA1基因未发生碱基突变。
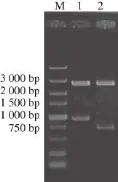
图3 克隆载体T-pMD19酶切鉴定Fig.3 Restriction enzyme digestion of recombinant cloning vector T-pMD19
2.2表达载体pY16TEF1-EGFP-APA1的构建
用XbaⅠ和EcoRⅠ、EcoRⅠ和ClaⅠ以及XbaⅠ和ClaⅠ分别酶切载体T-pMD19-APA1、T-pMD19-EGFP以及pY16,得到酶切产物,将酶切产物用T4连接酶连接,转化大肠杆菌DH5α,菌落PCR验证后,提取阳性克隆质粒,用XbaⅠ和ClaⅠ酶切验证,如图4所示。大片段大小约为5 500 bp左右,小片段约为1 700 bp左右,与预期相符。
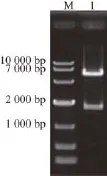
图4 表达载体pY16TEFF11--EEGGFFPP-AAPPAA11的酶切鉴定Fig.4 Restriction enzyme digestion of expression vector pY16TEF1-EGFP-APA1
2.3同义突变结果
以pY16TEF1-EGFP-APA1为出发载体,通过一步法,引入同义突变位点,扩增载体,得到带缺口的定点突变产物,用DpnⅠ处理产物,如图5所示。
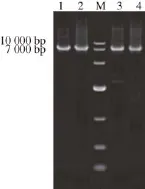
图5 质粒pY16TEFF11--EEGGFFPP-AAPPAA11的PPCCRR扩增Fig.5 PCR amplification of expression vector pY16TEF1-EGFP-APA1
酶切产物纯化后,转化大肠杆菌DH5α,菌落PCR验证,提取阳性克隆质粒送测序。测序结果表明成功引入同义突变,序列比对结果如图6所示。
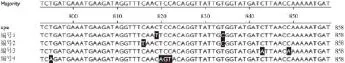
图6 阳性克隆质粒测序结果比对Fig.6 Comparison of DNA sequencing results
2.4APA1基因表达鉴定
用获得的同义突变载体转化酿酒酵母YS59感受态,挑取阳性克隆,菌落PCR验证后,YPD液体培养基培养,收集对数期细胞,荧光显微镜检测。如图7所示,镜检表明载体表达EGFP基因,因为该基因同目标基因APA1共表达,证明APA1基因表达。

图7 分别位于荧光(A)和普通光(B)下的酵母细胞Fig.7 Morphological observation of YS59 under fluorescence (A) and common light (B) microscope
3 讨 论
本研究基于一步法定点突变技术,该方法与QuickChangeTM以及重叠延伸法相比具有经济高效的特点。由于该方法的引物部分互补,使得一轮PCR后,扩增的片段可以作为下一轮反应的模板[23-24]。同时,引物部分互补,降低了引物形成二聚体的概率,因此更加高效。实验中,每次随机挑选4 个转化子测序,均成功引入突变。结合实际经验,一步法点突变技术中,模板浓度应尽量降低,过高的模板浓度会使得DpnⅠ不能彻底降解原始序列而增加假阳性。引物中突变位点应尽量远离引物两端,该位点可以位于互补区,也可以位于旁侧,一次可以引入两个突变位点。可以不需要购买QuickChangeTM试剂盒,用常规试剂就可以高效地实现定点突变。因此,合适的模板浓度以及彻底的DpnⅠ酶消化和合适的引物设计是该实验成功的关键。
将EGFP基因与目的基因共表达,通过荧光显微镜观察可以确定目的基因的表达以及表达强度,为获得不同表达量的基因奠定了基础。
4 结 论
测序结果与预期结果一致,成功获得了APA1的4 个同义突变APA1-1/2/3/4。并且这些同义突变转化酵母后,能与增强型绿色荧光蛋白共表达,为进一步不同表达量的APA1奠定了实验基础。一步法点突变技术是一种高效的定点突变方法。与QuickChangeTM相比,具有高效、经济的特点,而且可以一次引入两个位点。
[1]THOMAS D, SURDIN-KERJAN Y. Metabolism of sulfur amino acids in Saccharomyces cerevisiae[J]. Microbiology and Molecular Biology Reviews, 1997, 61(4): 503-532.
[2]马捷, 刘延琳. 葡萄酒中重要挥发性硫化物的代谢及基因调控[J].微生物学报, 2011, 51(1): 14-20.
[3]刘美玲, 刘延琳. 酿酒酵母单倍体的分离及其产硫化氢特性[J].食品科学, 2013, 34(21): 136-139. doi:10.7506/spkx1002-6630-201321028.
[4]LINDERHOLM A, DIETZEL K, HIRST M, et al. Identification of MET10-932 and characterization as an allele reducing hydrogen sulfide formation in wine strains of Saccharomyces cerevisiae[J]. Applied and Environmental Microbiology, 2010, 76(23): 7699-7707.
[5]LINDERHOLM A L, FINDLETON C L, KUMAR G, et al. Identification of genes affecting hydrogen sulfide formation in Saccharomyces cerevisiae[J]. Applied and Environmental Microbiology, 2008, 74(5): 1418-1427.
[6]BARTRA E, CASADO M, CARRO D, et al. Differential expression of thiamine biosynthetic genes in yeast strains with high and low production of hydrogen sulfide during wine fermentation[J]. Journal of Applied Microbiology, 2010, 109(1): 272-281.
[7]PLOTKIN J B, KUDLA G. Synonymous but not the same: the causes and consequences of codon bias[J]. Nature Reviews Genetics, 2011,12(1): 32-42.
[8]KUDLA G, MURRAY A W, TOLLERVEY D, et al. Coding-sequence dete rminants of gene expression in Escherichia coli[J]. Science, 2009,324: 255-258.
[9]QIAN Wenfeng, YANG Jianrong, PEARSON N M, et al. Balanced codon usage optimizes eukaryotic translational efficiency[J]. PLoS Genetics, 2012, 8(3): e1002603. doi: 10.1371/jo urnal.pgen.1002603.
[10] DING Y, SHAH P, PLOTKIN J B. Weak 5'-mRNA secondary structures in short eukaryotic genes[J]. Genome Biologyand Evolution, 2012, 4(10): 1046-1053.
[11] TULLER T, VEKSLER-LUBLINSKY I, GAZIT N, et al. Composite effects of gene determinants on the translation speed and density of ribosomes[J]. Genome Biology, 2011, 12(11): R110. doi:10.1186/gb-2011-12-11-r110.
[12] BENTELE K, SAFFERT P, RAUSCHER R, et al. Efficient translation initiation dictates codon usage at gene start[J]. Molecular Systems Biology, 2013, 9: 675. doi: 10.1038/msb.2013.32.
[13] LIU Huanting, NAISMIT H J H. An efficient one-step site-directed deletion,insertion, single and multiple-site plasmid mutagenesis protocol[J]. BMC Biotechnology, 2008, 8: 91. doi:10.1186/1472-6750-8-91.
[14] 张浩, 毛秉智. 定点突变技术的研究进展[J]. 免疫学杂志, 2009,16(4): 108-110.
[15] 周小玲, 沈微, 饶志明, 等. 一种快速提取真菌染色体DNA的方法[J].微生物学通报, 2004, 31(4): 89-92.
[16] 吴发红, 黄东益, 黄小龙, 等. 几种真菌DNA提取方法的比较[J]. 中国农学通报, 2009, 25(8): 62-64.
[17] 金筱耘, 赵爱春, 李军, 等. Monellin-EGFP融合蛋白在毕赤酵母中的表达[J]. 食品科学, 2012, 33(13): 171-175.
[18] 季爱加, 宁喜斌. 原核表达载体pET28a-EGFP的构建与表达[J]. 微生物学杂志, 2011, 31(4): 69-73.
[19]李利锋, 倪晔, 孙志浩. 利用定点突变法研究精氨酸脱亚胺酶活性的影响机制[J]. 生物工程学报, 2012, 28(4): 508-519.
[20] TSENG W C, LIN J W, WEI T Y, et al. A novel megaprimed and ligase-free, PCR-based, site-directed mutagenesis method[J]. Analytical Biochemistry, 2008, 375(2): 376-378.
[21] DANIEL GIETZ R, WOODS R A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method[J]. Methods in Enzymology, 2002, 350(1): 87-96.
[22] 王晓辉. 增强型绿色荧光蛋白在DH5α大肠杆菌中的表达[J]. 食品科学, 2010, 31(21): 200-203.
[23] DENG Q, LUO W, DONNENBERG M S. Rapid site-directed domain sca nning mutagenesis of enteropathogenic Escherichia coli espD[J]. Biological Procedures Online, 2007, 9(1): 18-26.
[24]戴灿, 苗聪秀, 卢光琇. 基于重叠延伸PCR法的定点突变技术[J]. 现代生物医学进展, 2010, 10(3): 411-412.
Site-Directed Mutagenesis of Saccharomyces cerevisiae APA1 and Co-expression with EGFP
MA Tao1, YAO Mingyue1, LIU Yanlin1,2,*
(1. College of Enology, Northwest A&F University, Yangling712100, China;2. Shaanxi Engineering Research Center for Wine and Viticulture, Yangling712100, China)
In order to further understand the relationship between the expression of APA1 and hydrogen sulfide production in Saccharomyces cerevisiae, synonymous mutations were introduced into APA1. The sequence of APA1 gene was cloned from S288c by PCR, connected with EGFP gene, and inserted into pY16 vector. The vector was used for synonymous mutations,and mutations of APA1-1/2/3/4 were obtained. According to the sequencing results, the mutations were correct. Synonymous mutations of APA1-1/2/3/4 were successfully acquired and could be used for further studies. Therefore, one-step sitedirected mutagenesis is an effective method. The fluorescence intensity of Saccharomyces cerevisiae YS59 transformed with pY16TEF1-EGFP-APA1-1/2/3/4 with fluorescence microscope confirmed that APA1 and EGFP were co-expressed.
vector construction; Saccharomyces cerevisiae; synonymous mutation
Q785
A
1002-6630(2015)23-0200-05
10.7506/spkx1002-6630-201523037
2015-01-31
国家自然科学基金面上项目(31571812);中央高校基本科研业务费专项资金项目(重点项目)(Z109021201);国家现代农业(葡萄)产业技术体系建设专项(CARS-30-jg-3)
马涛(1989—),男,硕士研究生,研究方向为葡萄酒微生物。E-mail:matao08@163.com
刘延琳(1966—),女,教授,博士,研究方向为葡萄酒及酿酒微生物。E-mail:yanlinliu@nwsuaf.edu.cn
猜你喜欢
环球时报(2022-09-20)2022-09-20
今日农业(2020年24期)2020-12-15
初中生学习指导·中考版(2020年7期)2020-09-10
西夏学(2019年1期)2019-02-10
西夏学(2018年2期)2018-05-15
中国调味品(2017年2期)2017-03-20
现代检验医学杂志(2016年5期)2016-08-20
兽医导刊(2016年12期)2016-05-17
中国科技信息(2015年2期)2015-11-16
现代检验医学杂志(2015年4期)2015-02-06
