
反相-高效液相色谱法同时检测食品中4种α-二羰基化合物
2015-10-21 08:18吴泰钢黄才欢白卫滨邱瑞霞张振华欧仕益
食品工业科技 2015年6期
吴泰钢,黄才欢,白卫滨,邱瑞霞,刘 柳,张振华,欧仕益
(暨南大学食品科学与工程系,广东广州510632)
反相-高效液相色谱法同时检测食品中4种α-二羰基化合物
吴泰钢,黄才欢,白卫滨,邱瑞霞,刘柳,张振华,欧仕益*
(暨南大学食品科学与工程系,广东广州510632)
建立了4种α-二羰基化合物[3-脱氧奥苏糖(3-DG)、乙二醛(GO)、丙酮醛(MGO)、2,3-丁二酮(2,3-BD)]的反相-高效液相色谱检测方法。首先将α-二羰基化合物与邻苯二胺反应生成喹喔啉衍生物(温度为60℃,pH=9.00,反应时间4h),然后用HPLC检测。色谱柱为Agilent ZORBAX SB-Aq(4.6mm×250mm,5μm),流速为0.7mL/min,流动相A为0.1%醋酸水溶液,B为甲醇,线性梯度洗脱程序为:0~35min,B 35%~70%;35~40min,B 70%~35%;40~50min,B 35%。30min内4种喹喔啉衍生物得到了较好的分离。方法学考察结果表明该HPLC方法具有很好的重现性和准确性,能够满足定量分析的要求。同时该方法适用于蜂蜜、食醋、凉果、咖啡中4种α-二羰基化合物含量的检测。
反相-高效液相色谱,α-二羰基化合物,邻苯二胺,测定
α-二羰基化合物(3-DG、GO、MGO、2,3-BD)是糖类通过焦糖化反应逆醛醇缩合和自动氧化[1]及Maillard反应过程中Amadori化合物降解形成的活泼中间产物[2],广泛存在于蜂蜜、啤酒、甜酒、咖啡、食用油、醋[1]、酸奶、酱油[3]中,是食品中风味和色泽物质的重要前体[4]。但摄入后会影响人体健康,如3-DG、GO、MGO会使蛋白质糖基化,诱导蛋白质形成糖化终末产物(AGEs)[5],从而导致细胞损伤,促使炎症的发生[6]。细胞中MGO和GO的浓度为1~2μmol/L,半数细胞毒性浓度为300μmol/L~1mmol/L[7]。2,3-BD容易导致人体患上乳糖不耐症以及肠道易激综合症[8]。因此,在常见加工食品中,探索准确、灵敏的α-二羰基化合物含量的检测方法,对于正确评价加工食品品质及安全性具有重要意义和应用价值。
α-二羰基化合物活性高、稳定性低,一般只能检测到天然能稳定存在的α-二羰基化合物或者经过还原反应后产生的醇类物质,因此难以满足对食品反应过程产生的所有α-二羰基化合物的定性及定量分析。目前,鉴定α-二羰基化合物的前处理方法通常是采用捕获剂法,即使用与α-二羰基化合物反应活性高的试剂,如巯基乙胺、氨基胍、甲基羟胺、Girard T试剂、邻苯二胺与α-二羰基化合物分别生成噻唑烷、3-氨基-1,2,4-三嗪衍生物、肟、脎、喹喔啉衍生物[2]。在所有的捕获剂中,邻苯二胺因为反应活性高,引起测量误差小,出现假阳性的概率低,因此是比较合适及常用的捕获剂[9]。
国内有报道关于二羰基化合物与糖尿病慢性并发症的关系[10],然而对于普通食品中这类化合物的检测方法尚未见报道。国外Ferreira,Daglia,Weigel[11-13]均采用邻苯二胺与α-二羰基化合物发生衍生反应从而检测它们在普通食品中的含量。然而两者的反应pH、时间、温度各异。因此本研究首先探讨了3-DG与邻苯二胺的最佳反应pH和时间,然后建立4种α-二羰基化合物的反相-高效液相色谱检测方法,并将此方法应用于蜂蜜、醋、凉果、咖啡中α-二羰基化合物含量的检测。期望公众对于食品中α-二羰基化合物的安全性引起高度的重视并为其含量的检测提供有效的参考。
1 材料与方法
1.1材料与仪器
3-DG(4084-27-9)、GO(107-22-2)、MGO(78-98-8)、2,3-BD(431-03-8)百灵威公司;邻苯二胺广州化学试剂公司;蜂蜜、咖啡、凉果、醋暨南大学兴安超市;甲醇(色谱纯)美国Mallinckrodt Baker公司;液相用水华润怡宝食品饮料(深圳)有限公司;0.45μm微孔滤膜天津津腾实验有限公司。
pH计、UV-9600紫外-可见分光光度计北京瑞利分析仪器公司;粉碎机广州大祥电子机械设备有限公司;XW-80A微型漩涡混合仪上海沪西分析仪器厂;DF-101S集热式恒温加热磁力搅拌器郑州英峪予华仪器有限公司;FM烘箱上海福玛实验设备有限公司;SHA-BA恒温振荡仪常州市伟嘉仪器制造有限公司;KDC-12低速离心机科大创新股份有限公司中佳分公司;LC-20AT高效液相色谱仪配备SPD-M 20A光电二极阵列检测器(PAD)和Lc-solution色谱工作站,日本岛津公司。
1.2喹喔啉衍生物生成的最佳条件
α-二羰基化合物因带双羰基结构具有高度的活性,4种α-二羰基化合物的结构式如图1所示,其与邻苯二胺反应生成喹喔啉衍生物,反应过程如图2所示,属于羰胺反应,受pH、温度和反应时间的影响。

图1 4种α-二羰基化合物的结构式Fig.1 Structural formular of 4α-dicarbonyls
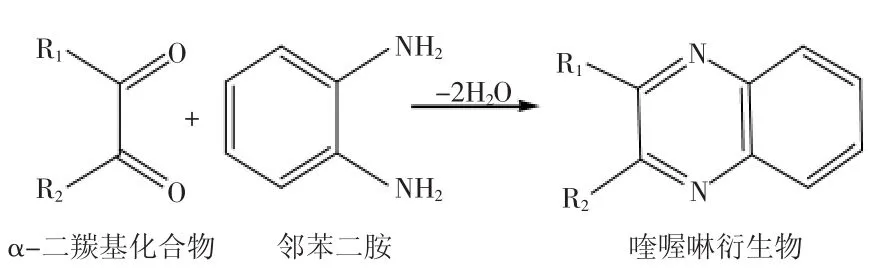
图2 α-二羰基化合物与邻苯二胺的反应过程[14]Fig.2 Derivatization reaction ofα-dicarbonyls with phenylenediamine[14]
3-DG是典型的α-二羰基化合物,因此,本研究用3-DG与邻苯二胺反应,探究喹喔啉衍生物生成的最佳pH和反应时间。
1.2.1喹喔啉衍生物生成的最佳pH向6支10.00m L试管中分别加入1m L 100μg/m L的3-DG溶液、1m L 1mg/m L邻苯二胺溶液,然后用0.1mol/L HCl溶液和0.1mol/L NaOH溶液将其pH分别调为1.60、2.90、3.70、6.80、9.00、10.00,最后用相应pH溶液定容至5.00m L。在60℃烘箱中反应1h,取样测量喹喔啉衍生物含量。
1.2.2喹喔啉衍生物生成的最佳反应时间向25.00m L试管中加入5.00m L 100μg/m L的3-DG溶液,5.00m L 1mg/m L邻苯二胺溶液,用0.1mol/L氢氧化钠溶液将其pH调为9.00,最后用相应pH溶液定容25.00m L,在60℃烘箱中反应1、2、3、4、5h,分别取样测量喹喔啉衍生物含量。
1.3样品液制备
1.3.1食醋样品测定取10.00m L醋,用0.1mol/L氢氧化钠溶液将其pH调为9.00,向其中加入0.10g邻苯二胺,用涡旋振荡器将其振荡均匀。然后将其置于恒温振荡器中反应4h(82r/min,温度60℃)。反应结束后,将样液在3500r/m in下离心5m in,取上清液用HPLC测定。
1.3.2蜂蜜样品测定取10.00m L蜂蜜,将其稀释6倍,其他操作和醋一样,取其上清液进行HPLC测量。
1.3.3凉果样品测定取凉果的果实将其放在105℃烘箱中烘干4h至恒重,用粉碎机粉碎,称10g粉末,加入100.00m L蒸馏水后,其他操作和醋一样,取其上清液进行HPLC测量。
1.3.4咖啡样品测定称10.00g咖啡粉末[12],加入100.00m L蒸馏水后,其他操作和醋一样,取其上清液进行HPLC测量。
1.4HPLC分析方法
色谱柱为Agilent ZORBAX SB-Aq(4.6mm×250mm,5μm),检测波长314nm,柱温40℃,进样量20μL,流速为0.7m L/m in,流动相A为0.1%醋酸水溶液,B为甲醇。梯度洗脱:0~35min,B 35%~70%;35~40min,B 70%~35%;40~50m in,B 35%。
1.5回收率以及仪器检出限和定量限的测定
回收率:向已知浓度的样品溶液中加入等体积的标准品溶液,浓度为20μg/m L,根据标准曲线,测定各物质含量。回收率(%)=100×(2×测定浓度-初始浓度)/加样浓度;仪器检出限(LOD)、定量限(LOQ)分别定义为3倍和10倍空白样品信号所对应的待测物浓度。
1.6数据处理方法
使用Excel处理数据,ChemDraw画化学式。所有定量结果测定3次,结果表示为平均值±标准偏差。
2 结果与分析
2.1喹喔啉衍生物生成的最佳pH
由图3可知,当pH=9.00时,3-DG与邻苯二胺反应生成喹喔琳衍生物的量最多。原因可能是在碱性环境下,邻苯二胺氨基去质子化,易于与3-DG发生反应,而pH太高则可能造成邻苯二胺脱氨[15]。
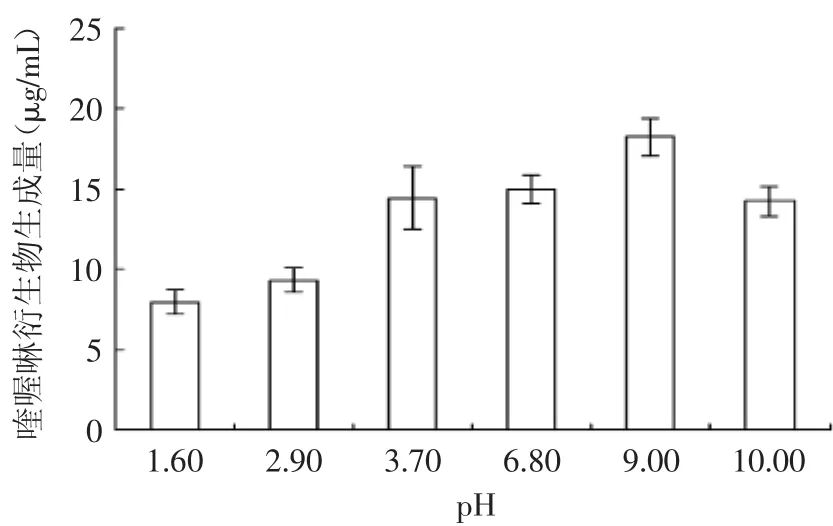
图3 pH对喹喔啉衍生物生成量的影响Fig.3 Effect of pH on the yield of quinoxaline derivative
2.2喹喔啉衍生物生成的最佳反应时间
由图4可知,3-DG与邻苯二胺反应前3h,喹喔啉衍生物生成量呈下降趋势,可能是前3h喹喔啉衍生物的消解大于生成。反应4h时,喹喔啉衍生物生成量最多。5h时,又呈下降趋势。因此,在60℃,pH=9.00条件下,3-DG与邻苯二胺最佳反应时间为4h。

图4 反应时间对喹喔啉衍生物生成量的影响Fig.4 Effect of time on the yield of quinoxaline derivative
2.3最佳检测波长的确定与分析
分别通过二极管阵列检测器扫描得到4种α-二羰基化合物标准品与邻苯二胺反应生成的喹喔啉衍生物紫外光谱图。扫描结果如图5所示。可以发现喹喔啉衍生物在190、230、314nm都出现吸收峰。然而,当检测波长为190和230nm时溶剂与其他杂质对目标物质干扰很大,因此最终检测波长确定为314nm。
2.4分离条件优化
在Arena[16]的方法的基础上进行优化,开始使用纯水和甲醇作为流动相,发现3-DG所对应的色谱峰出现不同程度的拖尾。向水中加入0.1%醋酸后,发现拖尾现象明显减小。可能因为3-DG属于多羟基化合物,在溶液中以离子态的形式存在,呈弱酸性,加醋酸抑制3-DG电离,从而改善其峰形。
采用流动相为甲醇和0.1%的醋酸溶液进行梯度洗脱时,发现当甲醇浓度在70m in之内从20%升高到36%,四种标准品分离度很差。当甲醇浓度在70min之内从30%升高到70%,4种标准品分离效果很好,但分离时间较长。将甲醇初始浓度调整为35%后,4种标准品不仅分离度很好,而且出峰时间明显缩短,30m in内4种标准品得到很好分离(图6),3-DG、GO、MGO、2,3-BD的保留时间分别为9.436、19.54、23.24、26.40m in。
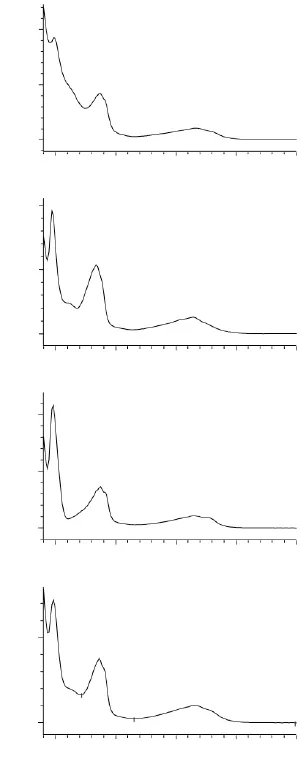
图5 喹喔啉衍生物紫外光谱图Fig.5 Ultraviolet spectra of quinoxaline derivative
2.5定量分析方法学考察[17]
建立了3-DG、GO、MGO、2,3-BD定量分析方法,方法学考察结果见表1。在线性范围内,均匀取6个浓度,测定其峰面积,以峰面积对浓度进行线性回归,得到标准曲线。各化合物线性关系良好,相关系数R2均大于0.994。方法精密度:连续测定浓度为20μg/m L的标准品3次,计算各标准品保留时间及峰面积的相对标准偏差。由表1可知,保留时间的相对标准偏差介于0.16%~0.65%之间,面积的相对标准偏差介于2.28%~4.42%之间,表明方法稳定性很好。

图6 α-DCC标准品(A)、醋与邻苯二胺反应生成的喹喔啉衍生物(B)色谱图Fig.6 The HPLC chromatography of quinoxaline derivative:standard(A)and sample(B)
测试结果显示:五种化合物的加标回收率介于97.3%~109.3%之间。4种化合物的仪器检出限(LOD)为0.01~0.42μg,定量限(LOQ)为0.04~0.89μg。方法学考察结果表明本HPLC方法具有良好的准确性、精密度、灵敏度,可用于实际样品的分析。
2.6样品分析
将建立的定量分析方法用于4种食品中α-二羰基化合物的定量分析,测定结果见表2。不同食品中α-二羰基化合物含量差异较大,4种食品中均检测到GO、MGO。3-DG在蜂蜜、凉果中含量远高于醋。醋中2,3-BD的含量是凉果的8.53倍。咖啡中3-DG、2,3-BD和蜂蜜中2,3-BD含量均低于检测限。
3 结论
在温度为60℃的条件下,3-DG与邻苯二胺衍生反应的最佳条件是pH=9.00,反应时间4h;色谱柱为ZORBAX SB-Aq(4.6mm×250mm,5μm),检测波长314nm,柱温40℃,进样量20μL,流速为0.7m L/m in,流动相A为0.1%醋酸水溶液,B为甲醇,线性梯度洗脱程序为:0~35min,B 35%~70%;35~40min,B 70%~35%;40~50m in,B 35%。30m in内4种喹喔啉衍生物得到了较好的分离。方法学考察结果表明该HPLC方法具有很好的重现性和准确性;3-DG在蜂蜜、凉果中含量远高于醋。醋中2,3-BD的含量是凉果的8.53倍。咖啡中3-DG、2,3-BD和蜂蜜中2,3-BD含量均低于检测限。

表1 检测方法线性、精密度及回收率实验结果Table 1 Precision,linearity and recovery of HPLCmethod of different analyzes
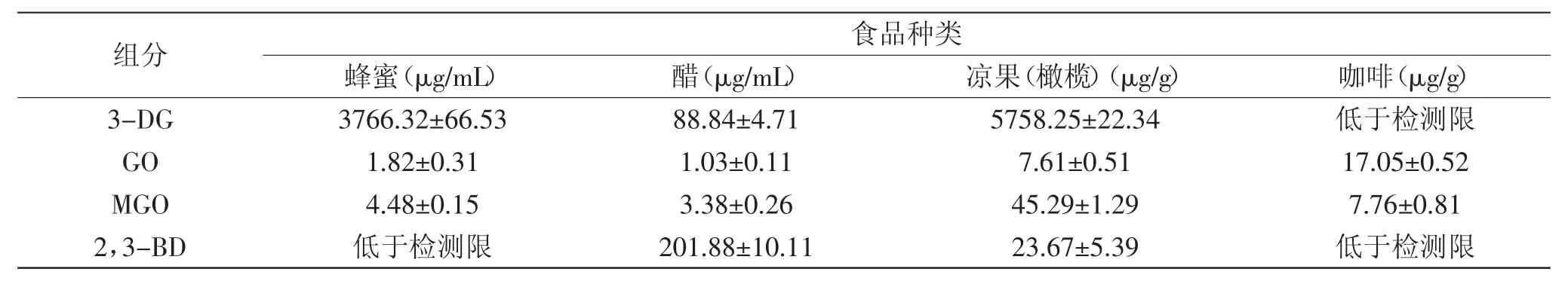
表2 4种食品中α-二羰基化合物含量Table 2 Contents ofα-dicarbonyls in four kinds of food
[1]Daglia M,Amoroso A,Rossi D,et al.Identification and quantification ofα-dicarbonyl compounds in balsamic and traditional balsamic vinegars and their cytotoxicity againsthuman cells[J].Journal of Food Composition and Analysis,2013,31:67-74.
[2]Chen SM,Kitts D D.Identification and quantification ofαdicarbonyl compounds produced in different sugar-amino acid Maillard reactionmodel systems[J].Food Research International,2011,44:2775-2782.
[3]Papetti A,Mascherpa D,Gazzani G.Freeα-dicarbonylcompounds in coffee,barley coffee and soy sauce and effects of in vitro digestion[J].Food Chemistry,2014,164:259-265.
[4]Glomb M A,Tschirnich R.Detection ofα-Dicarbonyl Compounds in Maillard Reaction Systems and in Vivo[J].Journal of Agricultural and Food Chemistry,2001,49:5543-5550.
[5]Lopez D R,Lecomte M,MoinetG,etal.Reaction ofmetformin with dicarbonyl compounds.possible implication in the inhibition of advanced glycation end product formatio[J].Biochemical Pharmacology,1999,58:1765-1773.
[6]Thornalley J.Dicarbonyl intermediates in theMaillard reaction[J].Annals New York Academy of Sciences,2005,1043:111-117.
[7]Darin D,Naila A,Song L J,et al.Increased dicarbonyl metabolisminendothelialcellsinincreaseddicarbonyl metabolism in endothelial cells[J].Diabetes,2006,55(7):1961-1969.
[8]Campbell A K,Matthews S B,Vassel N,et al.Bacterial metabolic‘toxins’:A new mechanism for lactose and food intolerance,and irritable bowel syndrome[J].Toxicology,2010,278:268-276.
[9]吕梦莎.牛肉香精的制备及Maillard反应中间产物—Alpha-二羰基化合物的研究[D].广州:暨南大学,2012.
[10]王倩,江国荣,王纯庠.二羰基化合物与糖尿病慢性并发症关系的研究进展[J].安徽医药,2010,14(2):130-132.
[11]Ferreira A C,Reis S,Rodrigues C,et al.Simultaneous determinationof ketoacids anddicarbonyl compounds,key Maillard intermediates on the generation of aged wine aroma[J]. Journal of Food Sciences,2007,72(5):314-318.
[12]Daglia M,Camillaaceti A,Sordelli B,et al.Isolation and Determination ofα-Dicarbonyl Compounds by RP-HPLC-2,3-BDD in Green and Roasted Coffee[J].Journal of Agricultural and Food Chemistry,2007,55:8877-8882.
[13]Weigel K U,Opitz T,Henle T.Studies on the occurrence and formation of 1,2-dicarbonyls in honey[J].European Food Research Technology,2004,218:147-151.
[14]Sabrina G,Mittelmaier S,Marcus A,et al.Identification and quantification of sixmajorα-dicarbonyl process contaminants in high-fructosecornsyrup[J].Analytical andBioanalytical Chemistry,2012,403:2923-2931.
[15]汪小兰.有机化学[M].第四版.北京:高等教育出版社,2005:161.
[16]Arena E,Ballistreri G,Tomaselli F,et al.Survey of 1,2-dicarbonyl compounds in commercial honey of different floral origin[J].Journal of Food Science,2011,76(8):1203-1210.
[17]张清峰,张汉扬,上官新晨,等.高效液相法同时测定土茯苓中五种多酚成分[J].现代食品科技,2013,29(9):2275-2279.
Simultaneous determ ination of fourα-dicarbonyls in food by RP-HPLC-DAD
WU Tai-gang,HUANG Cai-huan,BAIWei-bin,QIU Rui-xia,LIU Liu,ZHANG Zhen-hua,OU Shi-yi*
(Departmentof Food Science and Engineering,Ji’nan University,Guangzhou 510632,China)
ARP-HPLCquantitative methodfor simultaneous determ ination of fourα-dicarbonyls,3-deoxyg lucosulose(3-DG),g lyoxal(GO),methylg lyoxal(MGO),and 2,3-butaned ione(2,3-BD),was estab lished.α-d icarbonyls were reacted w ith phenylenediam ine to form quinoxaline derivative at 60℃,pH9.00 for 4h.The derivatives were separated on an Agilent column ZORBAX SB-Aq(4.6mm×250mm,5μm)using 0.1%acetic acid aqueous solution(A)and methanol(B)as mobile phase for the follow ing stepw ise grad ient elution:35%~70%(B)in 0~35m in,70%~35%(B)in 35~40m in,35%(B)in 40~50m in,the flow rate was 0.7m L/m in.The results showed that the four quinoxaline derivatives were well separated w ithin 30m in.This method could meet the quantitative analysis because of its good rep roducibility and accuracy confirmed by methodology.It was also well app lied in the determ ination of fourα-dicarbonyls in honey,vinegar,p reserved fruit,and coffee.
RP-HPLC;α-d icarbonyls;phenylened iam ine;determ ination
TS201.1
A
1002-0306(2015)06-0058-05
10.13386/j.issn1002-0306.2015.06.003
2014-06-19
吴泰钢(1990-),男,硕士研究生,研究方向:功能性食品。
欧仕益(1963-),男,博士,教授,研究方向:食品化学。
国家自然科学基金(31371745);科技部863项目(2012BAK01B03)。
猜你喜欢
中学生数理化·中考版(2021年12期)2021-12-31
中学生数理化·中考版(2021年11期)2021-12-06
陶瓷学报(2021年5期)2021-11-22
科学技术与工程(2021年8期)2021-04-22
陶瓷学报(2020年6期)2021-01-26
表面技术(2020年2期)2020-03-04
世界农药(2019年3期)2019-09-10
中学化学(2017年6期)2017-10-16
中学生数理化·高二版(2016年3期)2016-12-26
山东工业技术(2016年15期)2016-12-01
