
广西汉族早老症家系三例病例研究
——影像学特征分析
2015-09-14 03:12:50吴华裕覃霞舒艳张伟峰李福记舒伟马军胡启平袁志刚林有坤方玲
中国全科医学 2015年29期
吴华裕,覃霞,舒艳,张伟峰,李福记,舒伟,马军,胡启平,袁志刚,林有坤,方玲
·临床病例分析·
广西汉族早老症家系三例病例研究
——影像学特征分析
吴华裕,覃霞,舒艳,张伟峰,李福记,舒伟,马军,胡启平,袁志刚,林有坤,方玲
目的分析广西汉族1个罕见早老症家系3例患者的影像学资料,探讨该家系早老症患者的影像学特征。方法收集广西汉族1个早老症家系3例患者的临床资料及CT、X线、MRI影像学检查资料,对其影像学特点进行总结。结果先证者为7岁女孩,另2例患者分别为先证者的妹妹(3岁)和弟弟(1岁)。先证者颈部正侧位片示颅盖骨与下颌骨比例不协调,下颌骨发育不全,锁骨消失,梨形胸。3例患者双手正侧位片检查均显示双手指指关节屈曲畸形,骨质疏松;年长的2例患者双手正侧位片提示骨龄发育迟缓,指骨远端骨质溶解明显。3例患者肺部CT平扫结果均未提示肺部纤维化表现,但先证者胸廓及肺脏均小于正常儿童,且胸壁脂肪厚度较正常儿童薄,颅脑MRI显示脑颅骨比例增大,垂体大小正常。结论早老症患者具有特征性的影像学改变,包括骨质疏松症、小下颌、锁骨及指骨末端缺失变短、手指指关节畸形、梨形胸等,对该病的诊断和鉴别诊断具有重要意义。
衰老,过早;早老症;体层摄影术,X线;体层摄影术,螺旋计算机;磁共振成像
吴华裕,覃霞,舒艳,等.广西汉族早老症家系三例病例研究——影像学特征分析[J].中国全科医学,2015,18(29):3619-3623.[www.chinagp.net]
Wu HY,Qin X,Shu Y,et al.Study of three cases of hutchinson-gilford progeria syndrome in a Guangxi Han family: analysis of imaging feature[J].Chinese General Practice,2015,18(29):3619-3623.
早老症(Hutchinson-Gilford Progeria Syndrome,HGPS)最早于1886年被Hutchinson报道,是一种极为罕见的、以儿童过早老化为特征的疾病,通常由染色体1q21.2上LMNA基因的一个显性突变所致[1]。临床表现为全身复合性缺陷,以生长发育落后、衰老面容、过早脱发、骨骼异常为主要表现。多于1岁内起病,平均在13岁时有90%的患者由于加速的心血管粥样硬化导致心脑血管疾病而过早死亡[2]。HGPS根据临床特征差异分为典型HGPS与非典型HGPS。典型HGPS的诊断通常根据常见的早老特征进行识别,以高加索人群中的显性遗传病例为主要类型,患者LMNA基因的11号外显子发生点突变(G608G:1824C>T);非典型HGPS患者一般较典型HGPS患者表现出更为罕见的临床及影像学特征或累及的组织更广、病情更严重[3-4]。本研究分析我国广西汉族的1个常染色体隐性遗传的早老症家系,探讨该家系患者的影像学特征。
1 材料与方法
1.1 临床资料该早老症家系来自广西贺州市,家系两代共5名家庭成员。父母非近亲婚配,表型正常,子女3人均为早老症患者。先证者为7岁女孩,曾于2009年就诊于广西医科大学第一附属医院皮肤科,当时患者表现为明显的皮肤硬化、双下肢活动受限、皮肤色素沉着,但无典型早老面容。经血常规、生化、双手X线、肺部CT平扫、颅脑MRI等检查,诊断为儿童系统性硬皮病[5]。先证者的妹妹3岁,先证者的弟弟1岁,3人均为HGPS患者,发病过程基本一致,均为1岁内起病,表现为不同程度的生长发育缓慢、头发与眉毛稀少,甚至秃头,头皮及双下肢浅静脉显露,鸟形脸,小下颌、牙拥挤,全身皮下脂肪减少、四肢皮肤硬皮病样改变,指(趾)骨挛缩、关节活动度下降。先证者上述症状最严重,还存在斜颈;其妹妹症状次之,全部牙齿均为龋病;其弟弟症状最轻,仅表现有头发稀疏,双下肢皮肤肿胀,手关节稍屈曲畸形。3例患者智力和性格均无异常,均行肺部CT平扫和双手正侧位X线检查;先证者行颈部正侧位X线检查。
1.2 影像学检查仪器与方法采用Carestream CR或DR机摄颈部正侧位片和双手正侧位片;以GE 64排(Light speed VCT)螺旋CT机行肺部CT扫描,扫描参数为:电压为120 kV,电流200 mA,层厚5 mm,层距5 mm,扫描准值为0.75 mm,螺距1.5~1.8,重建层厚2 mm,FOV300 mm。以GE 1.5T超导MRI机行颅脑MRI扫描(仅用于先证者),头部线圈,仰卧位,分别用SE T1WI(1 000/10 ms),T2WI(3 000/ 80ms),DWI(6 000 ms/85 m,b值1 000 s/mm2),T2flair(8 400ms/120ms/2 100ms)序列扫描,FOV23 cm×23 cm,层厚6 mm,间隔2 mm,矩阵256×256。MRI增强:经肘静脉注射0.15 mmol/kg喷酸葡胺(Gd-DTPA),采用动态增强扰相梯度回波T1WI快速扫描序列进行扫描。对于不能配合检查的患儿检查前给予水合氯醛口服镇静。
2 结果
本研究3例患者双手正侧位X线检查均提示双手指指关节屈曲畸形和骨质疏松(见图1A、B、C),先证者2岁时双手正侧位X线检查已提示骨龄落后(见图1D),先证者妹妹亦存在骨龄落后(见图1B)。2例较年长患者指骨远端存在骨质溶解(见图1A、B)。先证者颈部正位片提示颅骨盖与下颌骨比例不协调,存在明显的下颌骨发育不良,小下颌,牙床拥挤畸形,脊柱向患者左侧弯曲,锁骨消失,胸廓呈梨形。颈部侧位片提示寰枕关节处发育不良,骨质密度异常增高(见图1E、F)。
3例肺部CT平扫结果均未提示肺部纤维化,但先证者胸廓存在畸形(脊突、椎体中点、胸骨中点并未在一条直线上),且大小较正常7岁女童(身高121 cm,体质量22 kg)小,在同一平面上(气管分叉处及心脏最大横径处)观肺脏及心脏大小亦均较正常7岁女童小,胸壁脂肪厚度较正常7岁女童稍薄(见图2)。先证者2岁时行胸部CT未见明显异常。颅脑MRI检查示先证者2岁时脑颅骨与面颅骨比例稍大,垂体大小正常,余未发现其他异常解剖结构及信号异常(见图3)。
3 讨论
3.1 背景HGPS是一种在儿童阶段即发生过早老化表现的疾病,至今为止全球范围内报道的病例约140例[6]。报道称90%以上的HGPS患者由定位于1q21.2上的LMNA基因的11号外显子发生点突变(G608G:GGC→GGT)引起,属于核纤层蛋白病[4],后发现R471C、R527C、G608S和C2036C>T等均与HGPS发病相关[7]。LMNA基因突变可导致至少8种遗传性疾病,包括肌肉性、神经源性、脂肪细胞变性方面疾病和衰老综合征,如Emery-Dreifuss肌肉萎缩症2型、肢带肌萎缩症1B型、扩张型心肌病1A型、Charcot-Marie-Tooth病2B1型、Dunnigan-type家族局部脂肪代谢障碍、下颌骨肢端发育不良1型(MADA)、限制性皮肤病(AD)和HGPS[8]。当发现LMNA基因突变位点相同,区别以上疾病时除了典型的临床表现外,影像学检查也是一项重要的检查措施,特征性的影像学改变可较容易地将HGPS与上述疾病区分。
对该家系3例患者行基因检测,发现该家系患者由LMNA基因第9号外显子上的R527C/R527C纯合突变所致[9],通过检索中国知网、万方、维普数据库及PubMed、Embase、Highwire外文数据库后发现这是目前国际上发现的第3个R527C纯合突变所致的HGPS家系,且3个HGPS家系均集中在我国[4,10]。HGPS患儿衰老极快,衰老速度相当于正常人群的5~10倍,这些患儿具有典型的临床表现,主要表现为严重生长迟缓、秃顶、皮下脂肪缺失、硬皮症、特征性颅面异常(如雕钩鼻、鸟样脸)、特征性骨改变(包括骨质疏松症、小下颌、锁骨的重吸收、指骨末端的吸收)、手指关节变硬、骑马样姿态、步态宽而拖曳、髋外翻、音调尖细,但其思维和认知能力正常,是一种部分衰老综合征[3,11]。本研究家系中3例患者均存在以上大部分临床特征,与典型的高加索人群HGPS患者临床表现类似。

图1 3例患者X线检查结果Figure 1 X-ray results of3 patients
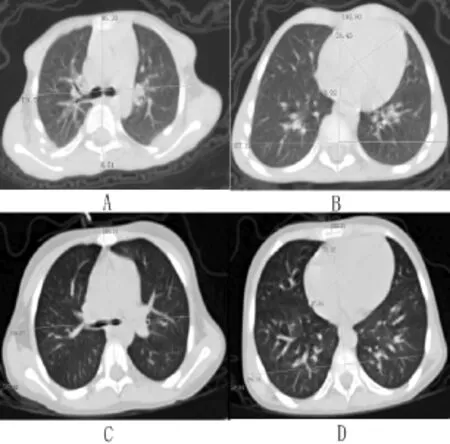
图2 先证者和正常同龄女童肺部CT检查Figure 2 Lung CT scanning results of the proband and a healthy girl at the same age
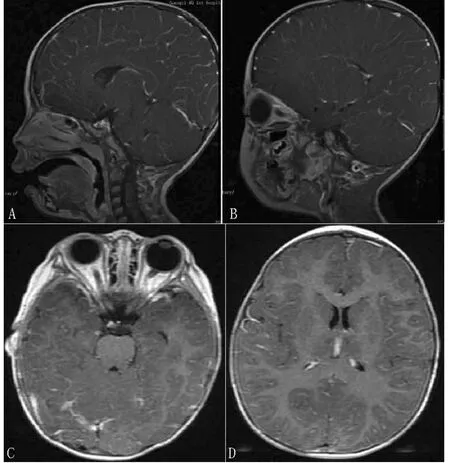
图3 先证者颅脑MRI检查结果Figure 3 Brain MRI imaging of the proband
3.2 HGPS的影像学特征HGPS患者的骨改变在影像学上有特征性表现。本研究发现患者双手指指关节屈曲畸形和骨质疏松、骨龄落后、指骨远端存在骨质溶解,脊柱侧弯,锁骨消失,梨形胸。经查阅文献发现大部分HGPS患者X线检查均可看到锁骨短小甚至缺失、骨质疏松、远端指(趾)骨骨质溶解吸收[4,6,12-14]。除以上这些改变外,有部分HGPS患者X线检查提示关节亦存在病变,部分表现为双侧髋外翻或髋关节脱位[12];Espandar等[15]报道的病例除髋关节脱位外,肩关节X线亦提示左前肩关节脱位;Akhbari等[16]报道的2例HGPS患儿中有1例存在双侧股骨头坏死。本研究3例患者双手多个指关节亦存在屈曲畸形。HGPS患者躯干、四肢骨骼特征性的影像学改变对诊断该疾病有重要意义。
本研究还发现先证者颈部正位片提示颅骨盖与下颌骨比例不协调,存在明显的下颌骨发育不良、小下颌、牙床拥挤畸形,颈部侧位片提示寰枕关节处发育不良、骨密度异常增高。同时颅脑MRI检查亦提示脑颅骨与面颅骨比例异常,但未发现垂体大小异常。这与大部分HGPS患者面颅骨及牙齿的典型影像学改变相符。Chen等[12]于2012年报道了1例8.5岁的中国HGPS女患儿颅骨X线提示小下颌、下颌骨发育不全所致的牙床拥挤,乳牙和恒牙同时存在,恒牙萌出延迟。Panigrahi等[13]2013年报道的HGPS患者亦提示小下颌,同时该患者患有严重龋病导致影像学上表现为部分牙齿不完整,缺齿。Reichert等[3]2014年报道的HGPS患者恒牙延迟萌出,且存在缺齿,磨牙间相嵌,咬合不全,颞下颌关节畸形,下颌关节CT检查表现为下颌关节窝被压扁,两边髁部发育不全。Kashyap等[6]报道的患者存在下颌骨前凸。Chen等[12]报道的患者MRI检查提示颅盖骨薄,脑颅骨与面颅骨比例较大,颅脑CT平扫提示上矢状窦附近的硬脑膜钙化。有研究报道HGPS患者颅脑CT提示蝶骨、额骨、上颌窦发育不全,部分患者骨缝未闭合,部分患者提示颅盖骨薄且相对宽、板障空间缺失或变浅[3,6,13];但也有报道称颅脑MRI提示脑垂体小[4],这与本研究家系先证者MRI结果不符。以上患者影像学上还提示梨形胸或严重的脊柱侧凸畸形。本研究家系患者除存在以上大部分特征性影像学上改变外,还存在寰枕关节处发育不良、骨密度异常增高,导致功能上旋转头部受阻。
从肺部CT结果看先证者胸廓大小较正常同龄女童小(梨形胸),相应的肺脏及心脏大小亦均较正常年龄女童小,胸壁脂肪厚度较正常7岁女童稍薄。从以上影像学结果中不难发现不同HGPS患者影像学表现不完全相同,其中一个原因可能是因本病患者症状随年龄增大而加重,另一个原因可能与突变位点的差异及地域差异有关。
3.3 探究影像学改变原因HGPS患者影像学改变主要为骨骼改变所致,Gordon等[17]曾对26例均由G608G突变所致的HGPS患儿和57例正常儿童进行骨骼方面的对比研究,结果发现在HGPS患儿中横向面积骨密度得分较正常儿童低,但经过身高和体质量校正后得分改进,而纵向松质骨容积骨密度变化不大,且HGPS患儿骨结构及骨强度与正常儿童相比存在明显差异。这些结果均是在进食量相同的前提下得出,这表明HGPS并不是一种因营养不良所致的骨缺失疾病。骨密度及骨结构、骨强度的改变在影像学上可以表现为骨质疏松、骨质溶解、骨骼畸形等,Gordon等[17]的结果提示HGPS是一种独特的骨骼发育不良疾病,同时也进一步验证了HGPS患儿影像学上的特征性改变。
3.4 HGPS预后HGPS是一种极其罕见且危害性较大的疾病,该病随年龄增大症状明显加重。本研究先证者2009年曾到本院皮肤科就诊,但由于患者早老症状不明显,所以误诊为儿童系统性硬皮病[5]。HGPS患儿多半于13岁左右死于心血管疾病。针对该病国际上尚无有效的药物,但有相当一部分研究表明法尼基化抑制剂(洛那法尼和替吡法尼)可以逆转G608G突变所致表达早老蛋白细胞的核形态改变,但该药物仅限于有早老蛋白堆积的疾病。目前一般推荐使用小剂量阿司匹林预防心血管疾病的发生,以延长寿命。故针对本病的早期诊断、早期预防性用药将对HGPS患儿有极大好处;特征性的影像学改变对该病的诊断至关重要,因根据典型的临床表现选择适当的影像学检查方法,尽量减少该病的漏诊、误诊。
伦理要求:参与试验的患者个体对试验过程完全知情同意,在充分了解本治疗方案的前提下签署“知情同意书”;干预及治疗方案获医院伦理委员会批准。
作者声明:文章为原创作品,数据准确,内容不涉及泄密,无一稿两投,无抄袭,无内容剽窃,无作者署名争议,无与他人课题以及专利技术的争执,内容真实,文责自负。
[1]Zhang H,Chen X,Guo Y,et al.Hutchinson-Gilford progeria syndrome:report of2 cases and a novel LMNA mutation of HGPS in China[J].JAm Acad Dermatol,2013,69(4):e175-176.
[2]Baek JH,Schmidt E,Viceconte N,et al.Expression of progerin in aging mouse brains reveals structural nuclear abnormalities without detectible significant alterations in gene expression,hippocampal stem cells or behavior[J].Hum Mol Genet,2015,24(5):1305-1321.
[3]Reichert C,Golz L,Gotz W,et al.Dental and craniofacial characteristics in a patient with Hutchinson-Gilford progeria syndrome[J].JOrofac Orthop,2014,75(4):251-263.
[4]Xiong Z,Lu Y,Xue J,et al.Hutchinson-Gilford progeria syndrome accompanied by severe skeletal abnormalities in two Chinese siblings:two case reports[J].J Med Case Rep,2013,7: 63.doi:10.1186/1752-1947-7-63.
[5]Luo YY,Lin YK,Cao CW.A case of diagnosis and treatment of Juvenile Systemic Sclerosis[J].Chin J Derm Venereo,2010,24 (9):850-851,870.(in Chinese)
罗彦彦,林有坤,曹存巍.儿童系统性硬皮病1例[J].中国皮肤病性病学杂志,2010,24(9):850-851,870.
[6]Kashyap S,Shanker V,Sharma N.Hutchinson-Gilford progeria syndrome:a rare case report[J].Indian Dermatol Online J,2014,5(4):478-481.
[7]Cao H,Hegele RA.LMNA ismutated in Hutchinson-Gilford progeria (MIM 176670)but not in Wiedemann-Rautenstrauch progeroid syndrome(MIM 264090)[J].Journal of Human Genetics,2003,48(5):271-274.
[8]Luo DQ,Wang XZ,Meng Y,et al.Mandibuloacral dysplasia typeA-associated progeria caused by homozygous LMNA mutation in a family from Southern China[J].BMC Pediatr,2014,14:256-263.
[9]Qin X,Luo YY,Yuan GZ,etal.Analysis of clinical characteristics and causative genes of Hutchinson-Gilford progeria syndrome in a family[J].Chin JDermatol,2015,48(3):184-186.(in Chinese)
覃霞,罗彦彦,袁广之,等.一个儿童早老症家系临床特征分析和致病基因研究[J].中华皮肤科杂志,2015,48(3):184-186.
[10]Liang L,Zhang H,Gu X.Homozygous LMNA mutation R527C in atypical Hutchinson-Gilford progeria syndrome:evidence for autosomal recessive inheritance[J].Acta Paediatr,2009,98 (8):1365-1368.
[11]Narazaki R,Makimura M,Sanefuji M,et al.Bilateral stenosis of carotid siphon in Hutchinson-Gilford progeria syndrome[J].Brain Dev,2013,35(7):690-693.
[12]Chen CP,Lin SP,Lin DS,etal.Clinical imaging findings in a girl with Hutchinson-Gilford progeria syndrome[J].Genet Couns,2012,23(1):1-7.
[13]Panigrahi RG,Panigrahi A,Vijayakumar P,et al.Hutchinson-Gilford progeria syndrome:a rare genetic disorder[J].Case Rep Dent,2013,2013:631378.
[14]Huang S,Liang Y,Wu W,et al.Analysis of a case with typical Hutchinson-Gilford progeria syndrome with scleroderma-like skin changes and review of literature[J].Chiese Journal of Pedlatrics,2014,52(2):112-116.
[15]Espandar R,Eraghi AS,Mardookhpour S.Simultaneous shoulder and hip dislocation in a 12-year-old girl with hutchinson-gilford progeria syndrome[J].Acta Med Iran,2012,50(6):439-443.
[16]Akhbari P,Jha S,James KD,et al.Hip pathology in Hutchinson-Gilford progeria syndrome:a reportof two children[J].JPediatr Orthop B,2012,21(6):563-596.
[17]Gordon CM,Gordon LB,Snyder BD,et al.Hutchinson-Gilford progeria is a skeletal dysplasia[J].JBone Miner Res,2011,26 (7):1670-1679.
Study of Three Cases of Hutchinson-Gilford Progeria Syndrome in A Guangxi Han Fam ily:Analysis of Imaging Feature
WU Hua-yu,QIN Xia,SHU Yan,et al.Department of Cell Biology and Genetics,Guangxi Medical University,Nanning 530021,China
Objective To study the imaging data of three cases of Hutchinson-Gilford Progeria Syndrome(HGPS) in Guangxi Han family,and to explore the patients'imaging features.Methods We collected the clinical data,CT,X-ray and MRIof three HGPS patients in Guangxiand made a summary of the imaging features.Results The proband was a 7-year-old girl,and the other two patients were her younger sister(3 years old)and brother(1 years old).The proband neck radiograph showed abnormal scale between the calvarium and the mandible,mandibular hypoplasia,clavicle disappearing and pear shaped chest.The X-ray of 3 patients'hands showed that all the 3 cases were with flexion deformity in digintal joints and osteoporosis,and two older patients showed retardation ofbone age and obvious osteolysis in distal phalanx.The lung CT scaning results of the 3 patients did not show lung fibrosis,but the chestand lung of the proband were smaller than normal children,and the fat thickness of the chest was thinner than normal girls.The brain MRI imaging of the proband showed the proportion of cerebral cranium became larger,but the pituitary size was normal.Conclusion The characteristic changes of the imaging occur in HGPS patients,including osteoporosis,mandibular hypoplasia,clavicle and distal phalanx disappearing and shortening,flexion deformity of digital joints and pear shaped chest.The findings have great significance on the diagnosis and differential diagnosis of progeria.
Aging,premature;Hutchinson-Gilford progeria syndrome;Tomography,X-ray;Tomography,spiral computed;Magnetic resonance imaging
R 441.9
A
10.3969/j.issn.1007-9572.2015.29.026
2015-03-10;
2015-07-10)
(本文编辑:赵跃翠)
广西自然科学基金资助项目(2014GXNSFAA118138);广西自然科学青年基金资助项目(2013GXNSFBA019143);国家自然科学基金资助项目(81260479)
530021广西南宁市,广西医科大学基础医学院细胞生物学与遗传学教研室(吴华裕,李福记,舒伟,马军,胡启平,袁志刚,方玲);广西医科大学第一附属医院皮肤性病科(覃霞,林有坤);浙江省湖州市妇幼保健院(舒艳,张伟峰)
方玲,530021广西南宁市,广西医科大学基础医学院细胞生物学与遗传学教研室;E-mail:ligefangling@sina.com。林有坤,530021广西南宁市,广西医科大学第一附属医院皮肤性病科;E-mail:linyoukun7@aliyun.com
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
疯狂英语·新阅版(2021年6期)2021-07-19 22:16:54
中国临床医学影像杂志(2019年4期)2019-06-18 10:55:04
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
第二课堂(课外活动版)(2015年3期)2015-10-21 19:41:19
中国医疗美容(2015年1期)2015-07-12 10:06:54
