
介质阻挡放电在原子光谱分析中应用研究的新进展
2015-09-11 07:06邓宇佳李成辉蒋小明侯贤灯
分析化学 2015年9期
邓宇佳 李成辉 蒋小明 侯贤灯
摘 要 近年来,介质阻挡放电在分析化学领域,尤其是在光谱分析技术中获得广泛的关注。本文综述了2011年至今介质阻挡放电在原子光谱分析中的应用,包括原子发射光谱分析、原子吸收光谱分析、化学蒸气发生在原子光谱分析进样中的应用等。
关键词 介质阻挡放电; 原子发射; 原子吸收; 化学蒸气发生; 微等离子体
1 引 言
介质阻挡放电(Dielectric barrier discharge,DBD),又称为无声放电,它结构简单、能耗低,可在常温常压下产生非平衡态等离子体(Plasma)。典型的DBD装置可分为平板型(图1a~c)和同轴型(图1d~f)。两个电极之间至少需要一个阻挡介质(如玻璃、石英、陶瓷或聚合物等),放电间隙为0.1~10 nm。中间的放电区域充满气压为10~100 Pa的工作气体(氩气、氦气、氮气或者空气)。当在电极的两端加102~103 V、频率101~102 Hz的高压交流电时,DBD会放电产生110 eV的电子[1],这些电子与周围的气体分子发生非弹性碰撞,可以激发或解离气体分子, 产生包含自由基、离子、原子和分子碎片等多种物质的等离子体。DBD等离子体是一种非平衡态、低温、瞬时气体放电形成的(微)等离子体。关于DBD等离子体的特性研究已有报道[24],通常认为,当击穿电压超过帕邢(Paschen) 击穿电压时,电极间的气体会被击穿而产生放电,大量随机分布的微放电就会出现在间隙中,发出接近蓝紫色的光。由于DBD在放电过程中会产生大量化学性质非常活跃的自由基和准分子,能够提供足够的能量将分析物中的待测元素原子化形成基态的自由原子,甚至再将基态的自由原子激发到更高的激发态,然后在去激发态的过程中产生其原子发射光谱信号。因此,在原子光谱分析中,DBD可以用作激发源、原子化器、诱导化学蒸气发生等方面。与传统的电感耦合等离子体(ICP)类似,DBD等离子体中的金属元素的发光机理多为如下过程:
当然,DBD中也存在离解过程和电离过程,如潘宁电离等。因此,DBD在原子光谱分析中获得广泛应用。
DBD长期以来一直被用作臭氧发生装置[5],目前已经广泛地应用在灭菌[6]、化合物合成[7,8]、废物去除/降解[9,10]等领域。由于DBD优异的放电性能以及结构简单、工作寿命长、能耗低等诸多优点,吸引了众多分析工作者的关注。DBD自从2002年被引入光谱分析领域[11], 十多年来,它在分析化学领域得到了长足的发展。
原子光谱作为元素检测最有效的分析技术,可选择性、高灵敏地检测众多金属和非金属元素,具有分析速度快、准确度和灵敏度高等优点,在元素的痕量分析、形态分析中占据着非常重要的位置。常用的等离子体(如ICP)具有很高的激发能力,其稳定性好、基体效应小、线性范围宽,被广泛地用作元素原子发射光谱的激发源,但是其体积大、能耗高等缺点给其应用带来一定的限制。介质阻挡放电微等离子体被认为是一种低耗、有效的激发源,在原子光谱分析中得到了应用[12~14]。将其应用于原子光谱分析仪中,用作原子化器/激发源,能大大降低仪器的体积与能耗,将为便携式、小型化、野外、实时在线分析提供原子光谱分析新工具。
Karanassios[15]和Yuan[16]等先后综述了各类微等离子体的性质,包括DBD等离子体在分析化学中的应用。Meyer等[17]着重从结构设计上出发,对DBD在分析化学中的应用作了较全面的综述,本课题组[18]也综述了DBD在原子光谱、化学发光、气相色谱检测器、质谱离子源、离子迁移色谱等分析方面的应用。近年来,DBD作质谱离子源[19]也备受关注。至今为止,DBD已被应用于原子发射光谱分析(AES)、原子吸收光谱分析(AAS)、原子荧光光谱分析(AFS)、质谱分析(MS)等领域。本文重点综述了2011年至今,DBD在原子发射光谱、原子吸收光谱、化学蒸气发生进样等领域的新进展。DBD在AFS中的应用近年来鲜有报道,而DBD在MS中的应用多涉及分子质谱分析,因此本文不包括DBD在AFS的应用;对2011年之前的相关研究和DBD-MS感兴趣的读者,可参阅文献[20~22]。
2 DBD在原子光谱分析中的应用
2.1 DBD在原子发射光谱中的应用
2.1.1 气体(化)进样AES分析 DBD在氩气的氛围下具有作为冷激发源激发气体小分子的能力。溶液(雾化)进样容易消耗DBD等离子体的能量,甚至使等离子体熄灭。因此,为避免水溶液进样,可选择蒸气进样方式,将气态小分子引入DBD中激发有利于产生原子发射光谱。事实证明,化学蒸气发生(Chemical vapor generation,CVG)是DBD-AES有效的进样手段,能够有效地将分析元素从样品溶液中分离出来,进样效率高,分离效果好,避免了样品基体的干扰,也减少了等离子体能量的消耗,测定结果有较好的灵敏度和检出限。气态小分子与DBD工作气体混合进入DBD装置中,易于实现连续测定和自动化。常见的化学蒸气发生有:氢化物发生(Hydride generation,HG)、光诱导化学蒸气发生(Photochemical vapor generation,PVG)、卤化物发生(Halide generation)、氧化物发生(Oxide generation)、螯合物发生(Chelate generation)、烷基化合物发生(Alkylation generation)以及冷蒸气发生(Cold vapor generation)等。作为DBD的进样手段,HG应用最为广泛,主要原因是其反应速度快,蒸气发生效率高。但如果气液分离效果不好,也还会带入少量水分,从而影响原子特征谱线的测定,并导致基线漂移。为了彻底防止伴生水蒸汽进入DBD,常需要加除水装置。另外,产生过多的氢气易引起DBD等离子体猝灭。而PVG不需要类似HG的还原剂等,不产生氢气,还具有装置简单、成本低、较为绿色等优点。但其应用元素范围有限,且蒸气发生效率低、速度慢、产物稳定性不好等因素制约了其进一步发展。尽管还有诸多化学蒸气发生方式,但是大部分反应条件比较苛刻。电热蒸发作为新型DBD进样方式,具有样品消耗少、进样效率高、分离效果好、装置简单、易于自动化等优点,应当具有较好的发展前景。
2011年, Abdul-Majeed等[23]研制了小型便携式的DBD等离子体芯片,并用SnCl2还原水样中的汞为蒸气后进样,通过测定汞的原子发射谱线强度对其进行定量分析(检出限LOD=2.8 μg/L,相对标准偏差RSD=3.5%)。芯片化的设计虽然牺牲了灵敏度和检出限,但仍能满足工业和环境监测的要求,为AES的集成化、小型化拓宽了思路。Zhu等[24]利用HG-DBD-AES法测定了砷(LOD=4.8 μg/L)。他们并未采用传统的平板或同轴式DBD,而是用铜线圈作为外电极,钨棒作为内电极。该设计具有简单、功耗低、耗气量小等优点,有望发展成为廉价、稳定、便携、特定元素专用的原子发射光谱仪。
利用PVG能有效地将分析物引入DBD中,既避免了硼氢化物发生体系引入氢气猝灭等离子体,又降低了水蒸气的带入量,提高了等离子体的稳定性。He等[25]采用PVG-AES技术,对疫苗中的硫柳汞进行了测定,检出限为0.17 μg/L,在20 μg/L浓度下相对标准偏差为1.9%,较为稳定。仅需使用甲酸对样品进行处理,避免了强氧化剂和还原剂的使用,是一种较为绿色的检测手段。Cai等[26]也利用PVG产生的羰基镍蒸气直接通入DBD激发,采用镍232.0 nm的特征原子光谱线,对人发、紫菜、水样中的Ni进行了测定,检出限达到了1.3 μg/L。对于FeCo等元素,也应当可以用类似的PVG-DBD-AES法进行测定。
此外,氧化蒸气发生为DBD检测卤素实际样品提供了可行性。 Yu等[27]将紫菜、食盐、西地碘含片等样品预先处理还原为碘化物,通过与双氧水反应生成碘蒸气的方式进样,气液分离后在DBD中激发,用CCD进行检测,线性范围为0.1~10 mg/L,检出限为0.03 mg/L。之后,该课题组又利用相似的方式构建了一个小型化系统,将Br
用Sn2+预还原为Br
,再通过MnO4氧化为溴蒸气,对环境水样中的进行了测定[28](LOD=0.014 mg/L)。他们采用氧化蒸气发生的方式,为非金属元素的DBD发射光谱法测定提供了新的思路。
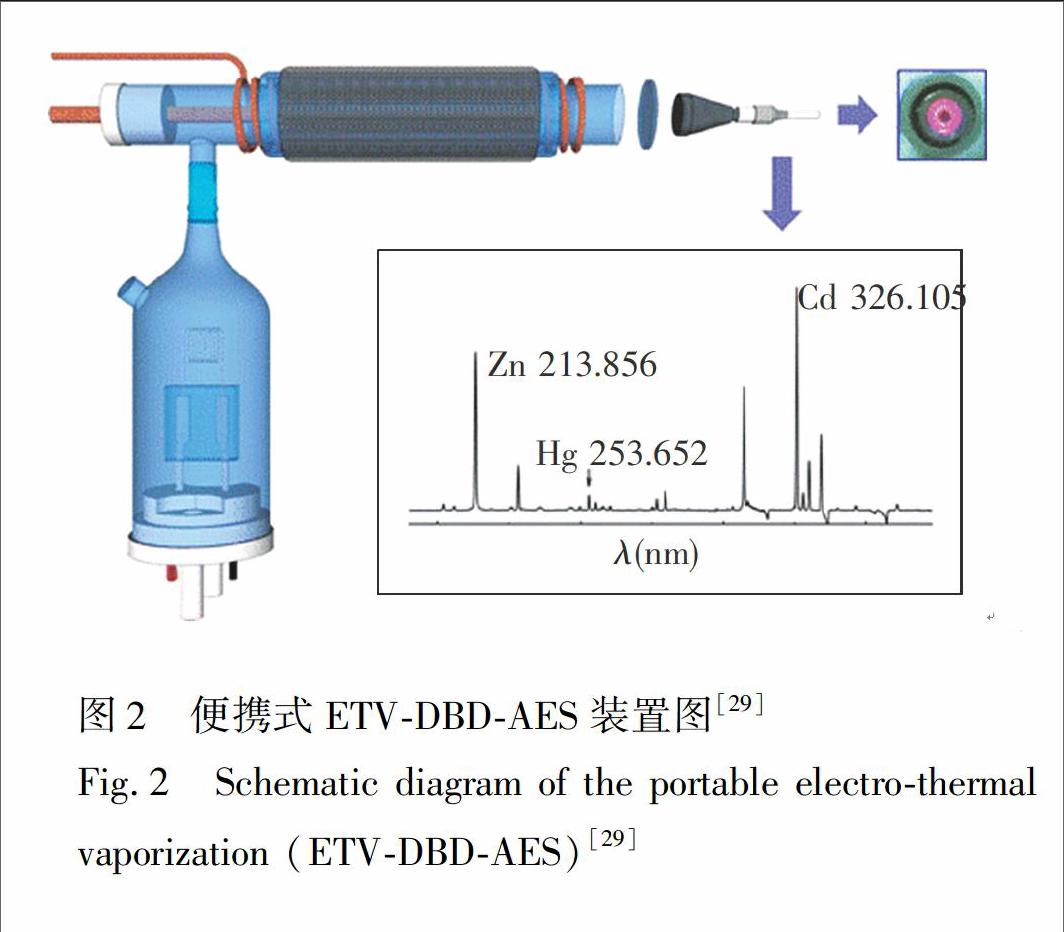
图2 便携式ETV-DBD-AES装置图[29]
Fig.2 Schematic diagram of the portable electro-thermal vaporization (ETV-DBD-AES)[29]
除化学蒸气发生外,电热蒸发(Electro-thermal vaporization, ETV)也是一种有效的样品引入技术。本课题组构建了钨丝电热原子化/蒸发-热辅助介质阻挡放电原子发射光谱分析系统(W-coil-ETV-DBD-AES)(图2)。将钨丝电热原子化器/蒸发装置作为进样装置和第一原子化器,能有效地通过升温程序去除水分和基体的干扰;另外,通过热辅助的方式可进一步确保DBD的激发能力。使用该装置对Cd 和Zn 进行了测定,结果表明, 该小型化仪器满足微量(10 μL)、快速(2 min)、灵敏的(LOD: Cd 0.8 μg/L, Zn 24 μg/L)分析要求[29]。 Zhu等也利用ETV进行微量(20 μL)进样,采用类似结构的DBD[30],对水样中的Pb进行了测定,从而进一步扩大了ETV-DBD-AES的应用范围。
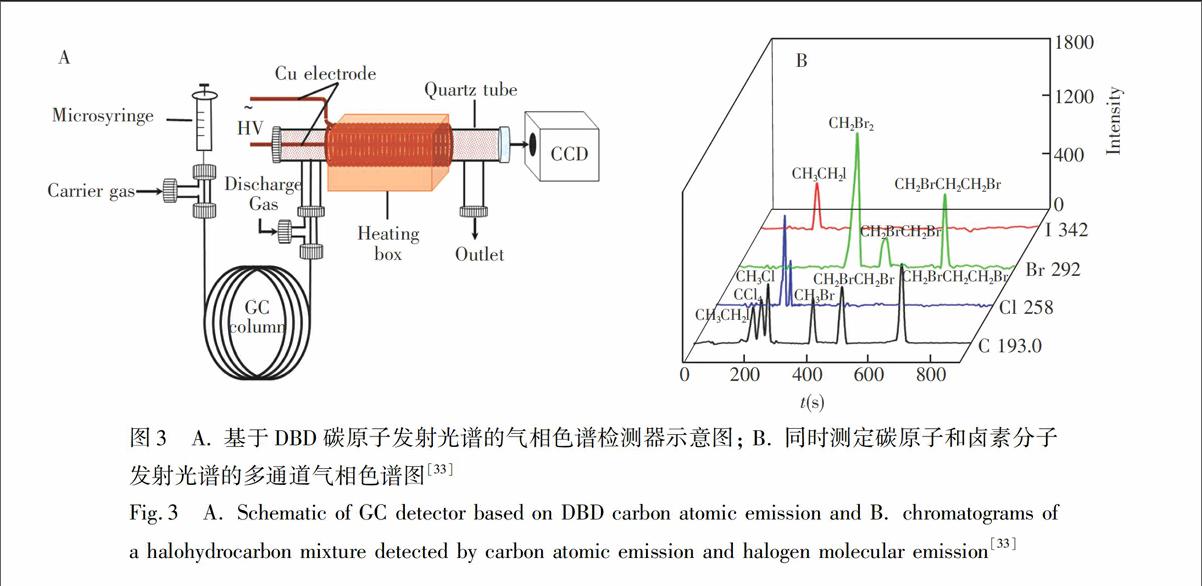
此外,本课题组将DBD作为气相色谱检测器,利用其发射光谱进行检测[31,32]。在色谱分辨率不足的情况下,利用其发射光谱信号的多通道同时检测,实现色谱-光谱多维信号分离,从而提高方法的分辨率,如图3。这种色谱检测器体积小、能耗低。非常有趣的是,DBD虽然能量低,但可激发碳原子产生193.7和247.8 nm
图3 A. 基于DBD碳原子发射光谱的气相色谱检测器示意图; B. 同时测定碳原子和卤素分子发射光谱的多通道气相色谱图[33]
Fig.3 A. Schematic of GC detector based on DBD carbon atomic emission and B. chromatograms of a halohydrocarbon mixture detected by carbon atomic emission and halogen molecular emission[33]的原子发射光谱[33],并可基于此构建广普型GC检测器用于含碳无机/有机化合物的(如图3B)检测。在此基础上,将微波辅助过硫酸钠湿法氧化与DBD相结合,借助于碳的原子发射光谱建立了在线的小型化的TOC(Total Organic Carbon)连续流动分析方法,并应用于环境水样的分析[34]。我们意识到,在DBD中碳的原子发射光谱的产生具有独特性;但遗憾的是,目前尚未有合适的机理可以解释这种现象。
2.1.2 液体进样AES分析 相比于气体分析,DBD对液体中金属元素的激发则更加困难。它需要更高的激发能力,也要降低溶剂对等离子体的干扰。Tombrink等[35]在DBD-AES液体分析方面做了大量研究工作。2010年,该课题组报道了一种特殊结构的DBD装置,在低液体流速条件下,直接用于溶液中的Na, Pb, Hg和Sr元素的原子发射光谱检测。由于此方法电极不接触溶液,减少了溶液对电极的氧化和腐蚀,延长了电极的使用寿命,并增加了放电的稳定性。在此基础上,增加液体流速,他们建立了液体电极介质阻挡放电(Liquid electrode dielectric barrier discharge, LE-DBD)装置,对溶液中的一些碱金属、碱土金属和银进行了测定[36]。以钨丝作为固体电极,含1 mol/L HNO3溶液作为液体电极,在石英毛细管中形成脉冲式放电,产生等离子体,对金属元素进行激发并检测其原子发射光谱。该装置充分发挥了原子发射光谱法多元素同时检测的优点,对包括碱金属、碱土金属、过渡金属、贫金属在内的23种元素进行了分析性能的评估,其中Li和Bi的检出限分别达到0.016 mg/L和41 mg/L[37]。
2012年, He等报道了基于一种液膜产生原子发射光谱的DBD装置[38],成功地激发了Na, K, Cu, Zn, Cd元素。该装置上部是一个钨丝电极,下方是一个拥有凹面的载玻片,载玻片下表面紧贴着一个铜片电极。将含1 mol/L的分析液体储存在凹面内,在两个电极加上高压交流电,直接产生等离子体。该装置有以下突出优点:不需要流动注射系统,直接移液到载玻片上进行分析;满足微量样品(小于80 μL)的分析需求等。因此,该设计可能拓展为高通量的阵列分析;由于其结构简单、成本低廉、低能耗, 以及无需供气系统等优点, 有望应用在现场分析领域。
2.2 作为原子吸收的原子化器
Kratzer等[39]将DBD作为原子化器应用于原子吸收光谱分析。他们利用二甲基二氯硅烷对DBD装置中的介质内表面进行甲硅烷基化,大大提高了Bi分析信号的强度,检出限为1.1 μg/L。推测其原因可能是: (1)未处理的介质表面与分析物有强烈的相互作用,导致了Bi原子沉积在介质内表面上,降低了信号强度; (2) 介质内表面处理过的DBD装置在含H2的Ar等离子体的作用下,能够有效移除残留在表面的Bi元素; (3) 不同于石英管原子化器(Quartz tube atomizer, QTA),氧气的存在会抑制信号的强度。此种DBD装置的准确度和精度可与QTA媲美,但灵敏度较差,检出限较低,可能是因为DBD原子化效率只有QTA的65%左右。可以预计,这种结构的DBD在元素的预富集方面存在潜在的应用价值。
2.3 用于等离子体诱导化学蒸气发生
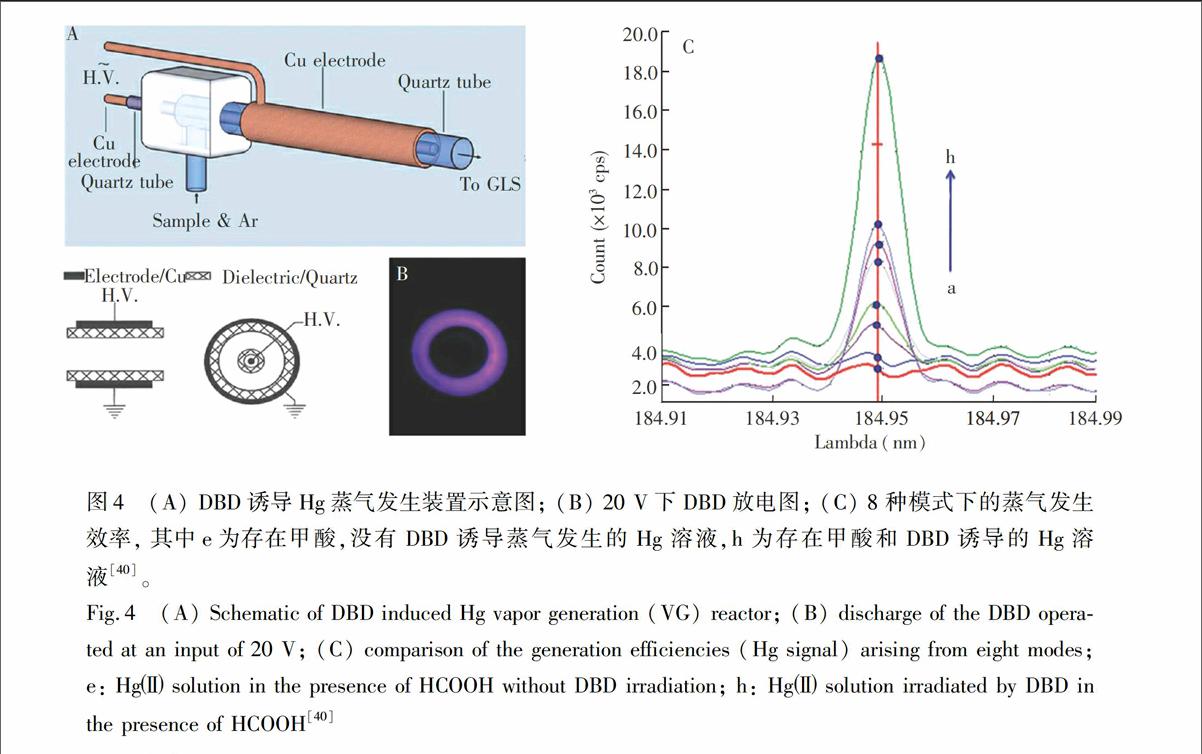
与液体阴极辉光放电(Electrolyte cathode glow discharge)诱导蒸气发生类似,DBD可以用于诱导蒸气发生。2011年,本课题组证实了DBD可用于Hg的诱导蒸气发生[40]。如图4所示,将液体以2 min通入DBD中进行放电,在没有其它化学物质加入的情况下,DBD能够诱导液体中的Hg原子化为Hg原子蒸气,通过气液分离后进入ICP-AES进行测定。如图4所示,甲酸能够明显增强Hg蒸气发生效率,检出限达到0.090 μg/L。将DBD诱导化学蒸气发生作为ICP-AES的进样方法,无需昂贵的雾化器,具有简单、灵敏、绿色等优点。基于DBD诱导化学蒸气发生,一种选择性测定痕量Se的新方法[41]得以建立(LOD=6 μg/L)。100 mg/L的Co2+, Ni2+, Sb3+, Ge2+和Zn2+对Se的测定无显著性影响,但低浓度的Cu2+对其有较大的影响。由于Se并不能通过这种方式进行蒸气发生,所以,该方法可望用于Se和Se的测定。
朱振利课题组在DBD诱导化学蒸气发生方面做了许多很好的研究工作。他们将外部包裹石英毛细管的铜电极插入待测溶液10 s取样后, 插入外表面包裹铝薄片的石英管中充当内电极,组成双介质阻挡放电装置。通电后产生Hg蒸气,进入原子荧光检测。利用该方法对水样、鱼肉样品中的汞、甲基汞和二甲基汞[42]、疫苗中的硫柳汞进行了测定[43]。由于是采用液体涂附在内电极的石英管外表面的进样方式,故只需消耗很少(大约6 μL)的样品即可完成测定。与对Hg的诱导蒸气发生不同,甲酸对信号的影响较小。
在此DBD装置的基础上,他们还通过定量流动注射的方式将液体从上而下通入DBD,形成一个液体薄膜,在线诱导Cd和Zn蒸气发生,最后用原子荧光光谱分析法进行测定[44,45]。在H2的辅助下,蒸气发生效率得到有效提高。其中,Cd的蒸气发生效率甚至优于HCl-KBH4-硫脲-Co2+体系,检出限达到0.03 μg/L(Ar-DBD)和0.008 μg/L(He-DBD);Zn的检出限也能达到0.2 μg/L,比常规的CVG技术改善了1020倍。
图4 (A) DBD诱导Hg蒸气发生装置示意图; (B) 20 V下DBD放电图; (C) 8种模式下的蒸气发生效率, 其中e为存在甲酸,没有DBD诱导蒸气发生的Hg溶液,h为存在甲酸和DBD诱导的Hg溶液[40]。
Fig.4 (A) Schematic of DBD induced Hg vapor generation (VG) reactor; (B) discharge of the DBD operated at an input of 20 V; (C) comparison of the generation efficiencies (Hg signal) arising from eight modes; e: Hg solution in the presence of HCOOH without DBD irradiation; h: Hg solution irradiated by DBD in the presence of HCOOH[40]
Yang等[46]证明了AsTeSb和Se可采用DBD产生的H2等离子体进行化学蒸气发生,从而代替传统的氢化物发生手段。他们首先将液体样品通过雾化器转化为气溶胶,在DBD等离子体中进行蒸气发生,最后通过原子荧光法进行测定,元素的绝对检出限分别为0.6, 1.0, 1.4和1.2 ng。其中,对于As的测定,他们结合了高效液相色谱(HPLC)分离,实现了As的形态分析,并且证明了AFS信号的提升来源于蒸气发生效率的提高,而与石英炉原子化器无关。
用于诱导蒸气发生的DBD装置, 从传统的同轴型结构,到液体薄膜-同轴双层结构,再到气溶胶-同轴结构,可以发现,液体与DBD等离子体充分接触是确保诱导蒸气发生的重要因素,并且H2在DBD诱导蒸气发生中起着重要的辅助作用。这也说明DBD诱导蒸气发生的能力还不够强,虽然H2和稀有气体的使用增加了诱导蒸气发生的成本,但其作为一种无需还原/氧化试剂的样品引入手段,具有运行简单、蒸气发生效率较高、低能耗、环境友好等特点, 有望应用在便携式光谱仪上。扩展DBD诱导化学蒸气发生的元素应用范围值也得我们进一步扩展相关研究。
3 结论和展望
由于DBD具有结构简单、能耗低、寿命长等优点,越来越受到野外分析和在线分析仪器研究者的青睐。DBD微等离子体已经成功实现了对气体样品和液体样品的原子分子光谱的激发,与此同时,发展能够与DBD装置联用的样品引入技术以及利用现有样品引入技术对测定元素进行扩展是DBD在光谱分析应用中的一个重要研究方向。目前,DBD微等离子体作为一种具有高能电子的低温等离子体,已经成功运用到了各类的小型化光谱分析系统中,实现了对多种元素或化合物的测定。多元素同时检测、高通量将是DBD的一个改进方向,对于更复杂样品的分析也将是一个巨大的挑战。我们相信,DBD因其独特优点,将在仪器小型化甚至微型化的应用中发挥着越来越重要的作用。
References
1 Snyder H R, Anderson G K. IEEE T. Plasma Sci., 1998, 26(6): 1695-1699
2 Tang J, Duan Y, Zhao W. Appl. Phys. Lett., 2010, 96(19): 191503
3 Shao T, Zhang C, Wang R, Zhou Y, Xie Q, Fang Z. IEEE T. Plasma Sci., 2015, 43(3): 726-732
4 Shao X J, Jiang N, Zhang G J, Cao Z X. Appl. Phys. Lett., 2012, 101(25): 253509
5 Kogelschatz U, Eliasson B, Egli W. Le Journal de Physique IV, 1997, 7(C4): 47-66
6 Pavlovich M J, Chang H W, Sakiyama Y, Clark D S, Graves D B. J. Phys. D: Appl. Phys., 2013, 46(14): 145202
7 Gómez-Ramírez A, Rico V J, Cotrino J, González-Elipe A R, Lambert R M. ACS Catal., 2014, 4(2): 402-408
8 Amorosi C, Fouquet T, Toniazzo V, Ruch D, Averous L, Ball V, Michel M. React. Funct. Polym., 2012, 72(5): 341-348
9 Zhang H, Li K, Sun T, Jia J, Lou Z, Feng L. Biochem. Eng. J., 2014, 241: 92-102
10 Hijosa-Valsero M, Molina R, Schikora H, Müller M, Bayona J M. J. Hazard. Mater., 2013, 262: 664-673
11 Miclea M, Kunze K, Franzke J, Niemax K. Spectrochim. Acta, Part B, 2002, 57(10): 1585-1592
12 YANG Meng, XUE Jiao, LI Ming, LI Jia, HUANG Xiu, XING Zhi. Chinese J. Anal. Chem., 2012, 40(8): 1164-1168
杨 萌,薛 蛟,李 铭,李 佳,黄 秀,邢 志. 分析化学, 2012, 40(8): 1164-1168
13 ZHANG Lu-Yuan, HU Sheng-Hong, LIU Yong-Sheng. Chem. J. Chinese Universities, 2008, 29(10): 1947-1952
张路远, 胡圣虹, 刘勇胜. 高等学校化学学报, 2008, 29(10): 1947-1952
14 DONG Li-Fang, SHANG Jie, JI Ya-Fei, LIU Liang, LI Xin-Chun. Spectroscopy and Spectral Analysis, 2012, 32(2): 302-305
董丽芳, 商 洁, 嵇亚飞, 刘 亮, 李新春. 光谱学与光谱分析, 2012, 32(2): 302-305
15 Karanassios V. Spectrochim. Acta, Part B, 2004, 59(7): 909-928
16 Yuan X, Tang J, Duan Y. Appl. Spectrosc. Rev., 2011, 46(7): 581-605
17 Meyer C, Müller S, Gurevich E, Franzke J. Analyst, 2011, 136(12): 2427-2440
18 Hu J, Li W, Zheng C, Hou X. Appl. Spectrosc. rev., 2011, 46(5): 368-387
19 Guo C, Tang F, Chen J, Wang X, Zhang S, Zhang X. Anal. Bioanal. Chem., 2014, 407(9): 2345-2364
20 Na N, Zhao M, Zhang S, Yang C, Zhang X. J. Am. Soc. Mass Spectrom., 2007, 18(10): 1859-1862
21 Venter A, Nefliu M, Graham Cooks R. TrAC, Trends Anal. Chem., 2008, 27(4): 284-290
22 Liu Y, Ma X, Lin Z, He M, Han G, Yang C, Xing Z, Zhang S, Zhang X. Angew. Chem. Int. Ed., 2010, 49(26): 4435-4437
23 Abdul-Majeed W S, Parada J H L, Zimmerman W B. Anal. Bioanal. Chem., 2011, 401(9): 2713-2722
24 Zhu Z, He H, He D, Zheng H, Zhang C, Hu S. Talanta, 2014, 122: 234-239
25 He H, Zhu Z, Zheng H, Xiao Q, Jin L, Hu S. Microchem. J., 2012, 104: 7-11
26 Cai Y, Li S, Dou S, Yu Y, Wang J. Anal. Chem., 2015, 87(2): 1366-1372
27 Yu Y L, Dou S, Chen M L, Wang J H. Analyst, 2013, 138(6): 1719-1725
28 Yu Y L, Cai Y, Chen M L, Wang J H. Anal. Chim. Acta, 2014, 809: 30-36
29 Jiang X, Chen Y, Zheng C, Hou X. Anal. Chem., 2014, 86(11): 5220-5224
30 Zheng H, Ma J, Zhu Z, Tang Z, Hu S. Talanta, 2015, 132: 106-111
31 Li W, Zheng C, Fan G, Tang L, Xu K, Lv Y, Hou X. Anal. Chem., 2011, 83(13): 5050-5055
32 Li C, Jiang X, Hou X. Microchem. J., 2015, 119: 108-113
33 Han B, Jiang X, Hou X, Zheng C. Anal. Chem., 2014, 86(1): 936-942
34 Han B, Jiang X, Hou X, Zheng C. Anal. Chem., 2014, 86(13): 6214-6219
35 Tombrink S, Muller S, Heming R, Michels A, Lampen P, Franzke J. Anal. Bioanal. Chem., 2010, 397(7): 2917-2922
36 Krhling T, Müller S, Meyer C, Stark A-K, Franzke J. J. Anal. At. Spectrom., 2011, 26(10): 1974-1978
37 Krahling T, Michels A, Geisler S, Florek S, Franzke J. Anal. Chem., 2014, 86(12): 5822-5828
38 He Q, Zhu Z, Hu S, Zheng H, Jin L. Anal. Chem., 2012, 84(9): 4179-4184
39 Kratzer J, Bousek J, Sturgeon R E, Mester Z, Dedina J. Anal. Chem., 2014, 86(19): 9620-9625
40 Wu X, Yang W, Liu M, Hou X, Zheng C, J. Anal. At. Spectrom., 2011, 26(6): 1204-1209
41 Yang W, Zhu X, Wu X. Chem. Res. Appl., 2011, 23(5): 644-648
42 Liu Z, Zhu Z, Wu Q, Hu S, Zheng H. Analyst, 2011, 136(21): 4539-4544
43 Wu Q, Zhu Z, Liu Z, Zheng H, Hu S, Li L. J. Anal. At. Spectrom., 2012, 27(3): 496
44 Zhu Z, Wu Q, Liu Z, Liu L, Zheng H, Hu S. Anal. Chem., 2013, 85(8): 4150-4156
45 Zhu Z, Liu L, Li Y, Peng H, Liu Z, Guo W, Hu S. Anal. Bioanal. Chem., 2014, 406(29): 7523-7531
46 Yang M, Xue J, Li M, Han G, Xing Z, Zhang S, Zhang X. Talanta, 2014, 126: 1-7
