
汞在MnOx-CeO2/γ-Al2O3催化剂表面的赋存形态分析
2015-08-21 07:02罗小雨苏胜向军汪一王鹏鹰尤默陆骑
化工学报 2015年6期
罗小雨,苏胜,向军,汪一,王鹏鹰,尤默,陆骑
(华中科技大学国家煤燃烧重点实验室,湖北 武汉 430074)
引 言
汞由于其易挥发性、生物累积性、剧毒性及可在全球范围内传播的特点,受到了人们的广泛关注。美国、加拿大、中国等国家已经制定了严格的法律法规来限制燃煤电站汞的排放。
燃煤烟气中汞的存在形式主要为单质汞(Hg0)、二价汞(Hg2+) 和颗粒态汞(Hgp)。Hg0熔点低、易挥发且难溶于水,与二价汞相比更难从烟气中除去[1]。在本课题组前期的研究实验中,使用溶胶浸渍法制备的MnOx-CeO2/γ-Al2O3(MnCe15)催化剂在固定床实验台架上展示了优良的对Hg0催化氧化性能[2]。在模拟烟气、SCR 操作温度条件下,MnCe15 催化剂可以达到超过80%的脱汞效率。为研究催化剂上汞与烟气组分之间可能的反应路径,实验考察了不同烟气组分(HCl、NO、O2)对催化剂上汞的催化氧化的影响,根据实验结果,HgCl2和HgO 被认为是极有可能的反应产物,并随着催化剂对二价汞的吸附逐渐饱和被排放进入尾部烟气中。
汞与HCl 在MnCe15 催化剂上的氧化可能遵循Langmuir-Hinshelwood 机理[2-4],即HCl 首先吸附在催化剂表面,被活化成为活性Cl,进而与邻近吸附位上的Hg0发生反应,生成HgCl2。Langmuir-Hinshelwood机理如图1所示。具体反应过程如下
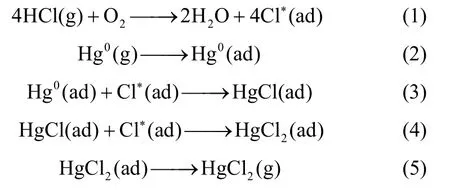
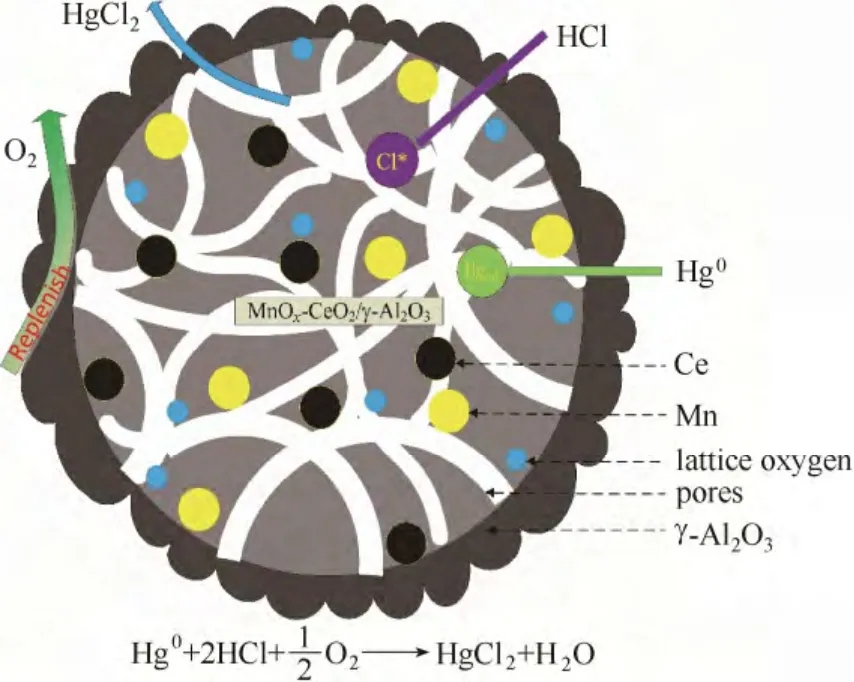
图1 Langmuir-Hinshelwood 反应机理Fig.1 Schematic diagram of Langmuir-Hinshelwood reaction mechanism
然而,并没有直接的实验证据证明这两种汞化合物HgCl2和HgO 真实存在于催化剂表面。这是由于吸附量过小,不同的汞化合物很难直接通过检测仪器进行测定、区分。
如果能够通过一定的方法证明在模拟烟气、SCR 操作温度下处理MnCe15 催化剂后存在相应的反应生成物HgCl2和HgO,则不仅可以证明“汞与HCl 在MnCe15 催化剂上的氧化遵循Langmuir- Hinshelwood 机理”这一论点的正确性,还可通过相同实验手法获得反应生成的其他汞化合物的实验数据,给出这些汞化合物真实存在于催化剂表面的直接证据。
利用不同的汞化合物在不同温度下的蒸气压相异这一特点,程序升温热分解(temperature- programmed thermal decomposition,TPTD)方法可以有效地区分、辨别不同的汞化合物,即在升温过程中汞化合物会在不同温度下分解,产生的挥发性的Hg0可以被后续的VM3000 在线测汞仪持续检测[5-6]。这一方法首先应用于研究岩石样品中的汞化合物[7]。随着测试技术的发展,TPTD 技术分别应用于研究氯碱化工厂和金矿附近受到汞污染的土壤样品[8]、汞矿附近受污染的水系样品[9]以及废弃荧光灯中汞的存在形式[10]。Uddin 等[11-14]研究了Fe2O3、Fe2O3/TiO2、FeS2和活性炭吸附剂在整体煤气化联合循环(IGCC)烟气和传统燃煤烟气条件下的脱汞效果,并使用TPTD 方法研究了吸附剂上汞化合物的形态。Lopez-Anton 等[15-21]将TPTD 方法应用于检测燃煤电站固体产物(如飞灰等)中富含的汞的形态。
目前为止,很少有使用TPTD 方法研究SCR 催化剂上汞的赋存形态的研究报道。因此,本工作的目的是使用MnCe15 催化剂,应用TPTD 方法对反应后MnCe15 催化剂的脱附曲线进行对比分析,研究其表面的汞化合物形态,并为进一步了解汞的氧化机理提供实验基础。
1 实验方法
1.1 催化剂的制备
本工作研究的MnCe15 催化剂采用溶胶-凝胶法制备,具体制备方法如下。
(1)向三口烧瓶中注入适量的去离子水,并在油浴锅中加热至85℃。
(2)去离子水温度保持85℃一段时间后,将称取的适量的异丙醇铝缓缓加入三口烧瓶中,加入异丙醇铝的同时打开电动搅拌器进行搅拌,防止发生凝结。
(3)搅拌过程中,混合液对空蒸发1 h,然后滴入适量浓度的硝酸,异丙醇铝逐渐水解变成浅白近透明的液体。调整油浴锅温度,使三口烧瓶中的温度升到95℃,并连接回流冷凝器,回流冷凝12 h。
(4)取下回流冷凝管,将溶胶对空蒸发2 h,以挥发溶胶中的酒精,所得即为勃姆石溶胶。
(5)将油浴温度调到70~75℃,待溶胶温度稳定后加入一定浓度的硝酸锰和硝酸铈溶液,所加入的硝酸锰和硝酸铈的量根据MnOx、CeO2质量负载量以Mn/(Mn+Ce)摩尔比为0.4 计算获得。边加边搅拌,直到黏度增大到搅不动为止。至此,溶胶转变成半凝胶。
(6)将体积比为10%的氨水注入到胶凝池中,深约10 cm。再加入深约20 cm 的液体石蜡。
(7)让溶胶老化10 min,用点滴器将直径2~3 mm 的半凝胶液滴滴入液体石蜡。溶胶边沉淀边收缩成球状,穿过石蜡与氨水的分界面,沉入氨水中老化。
(8)凝胶颗粒老化120 min 后取出。
(9)冲洗后,凝胶颗粒在真空干燥箱中干燥48 h。干凝胶呈深褐色球状,粒径为1~2 mm。最后在马弗炉中于550℃焙烧6 h。取出后在空气中冷却,即可得到催化剂。
1.2 实验系统
本研究在固定床实验台架上进行催化剂上汞的赋存形态分析。实验台架如图2所示,主要由高纯氮、程序升温反应炉和VM3000 在线测汞仪组成。
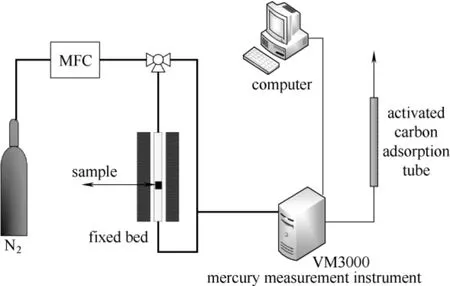
图2 TPTD 实验固定床反应系统Fig.2 Fixed bed reaction system of experiments
VM3000 测汞仪由德国Mercury Instruments 公司生产,采用原子吸收光谱法(AAS),可以连续在线监测气体中元素态汞的浓度,响应时间为1 s。为降低实验误差,本研究均采用较高的汞浓度(75 μg·m-3)。
1.3 实验过程
实验首先确定纯汞化合物的TPTD 曲线,并记录结果,作为对比组实验数据。为获得更加合理有效的实验数据,使用0.3 g新鲜的MnCe15催化剂(汞含量为0)与1 mg 不同的汞化合物混合,来模拟真实情况下不同汞物质在催化剂表面的分解行为。本研究使用纯的HgCl2、HgO 和HgSO4化合物,与催化剂混合进行实验。
为研究Hg0催化氧化反应后的MnCe15 催化剂上汞的赋存形态,本研究设计了多组预处理实验。实验使用0.3 g 催化剂在250℃条件下对催化剂进行预处理12 h,使得反应充分进行。预处理的烟气组分分别为:①Hg0;②Hg0+O2;③Hg0+HCl;④Hg0+HCl+O2;⑤Hg0+SO2+O2;⑥Hg0+NO;⑦Hg0+NO+O2;⑧Hg0+模拟烟气。将预处理工况下的催化剂记为Cx(x代表相应的烟气组分,例如C1)。具体实验工况见表1。整个实验过程中Hg0的浓度为75 μg·m-3,使用CO2作为载气,模拟烟气的组成成分为5% O2、10% CO2、0.001% HCl、0.02% NO、0.04% SO2、8% H2O,并使用氮气作为平衡气。表1中预处理使用N2作为平衡气,“—”代表没有此种气体。

表1 MnCe15 催化剂预处理实验工况Table 1 Experiment condition of MnCe15 catalyst pretreatment
将预处理后的MnCe15 催化剂置于程序升温反应炉中,使用流量为250 ml·min-1的高纯氮作为载气。为保证各种汞化合物的TPTD 曲线之间不出现相互重叠、覆盖,从而影响区分,本节以10℃·min-1的升温速率将温度由室温升至650℃。使用VM3000 测汞仪在线连续监测升温过程中汞化合物的分解情况。
2 实验结果与分析
新鲜MnCe15 催化剂分别与HgO、HgCl2和HgSO4混合后的TPTD 曲线如图3所示。由图3可以发现如下特征。

图3 纯汞化合物的TPTD 曲线Fig.3 TPTD curve of pure mercury compounds
(1)新鲜的MnCe15 催化剂在程序升温热分 解过程中并未观察到明显的Hg0脱附峰,这表明催化剂本身并不含汞。同时也证明在接下来的实验中出现的Hg0脱附峰来自纯汞化合物的分解。
(2)对于HgCl2,可以看到在50~160℃范 围内出现了一个较尖锐的Hg0脱附峰,峰值位于(100±3)℃处。
(3)HgO 的程序升温热分解发生在350~500℃范围内,峰值位于(405±3)℃处。
(4)在500~600℃温度范围内,HgSO4发生程序升温热分解行为,产生Hg0脱附峰,峰值位于(533±3)℃。
在C1 和C2 烟气条件下预处理过的MnCe15 催化剂的热分解曲线结果如图4所示。由图可以发现只有一个单独的Hg0脱附峰出现在400℃附近。与图3中的结果对比后可以发现,HgO 应该是主要存在于催化剂表面的物质,即在Hg0或Hg0+O2条件下MnCe15 催化剂可以将Hg0催化氧化为HgO。同时,由于O2的加入可以补充催化剂表面被消耗的活性氧,促进催化剂对Hg0的催化氧化[2],图4(b)中Hg0+O2的峰面积要大于图4(a)中Hg0单独存在时的峰面积,即Hg0+O2存在的条件下催化剂表面更多的Hg0被氧化为HgO。
C3 催化剂和C4 催化剂的程序升温热分解曲线如图5所示。Hg0+HCl 预处理后的C3 催化剂在约80℃和100℃处出现了两个Hg0脱附峰。与前人的研究结果[22-23]对比后可以发现,C3 催化剂表面Hg2Cl2和HgCl2共同存在,即只有HCl 存在的情况下会有部分Hg0被氧化为Hg2Cl2。可能的反应方程式为

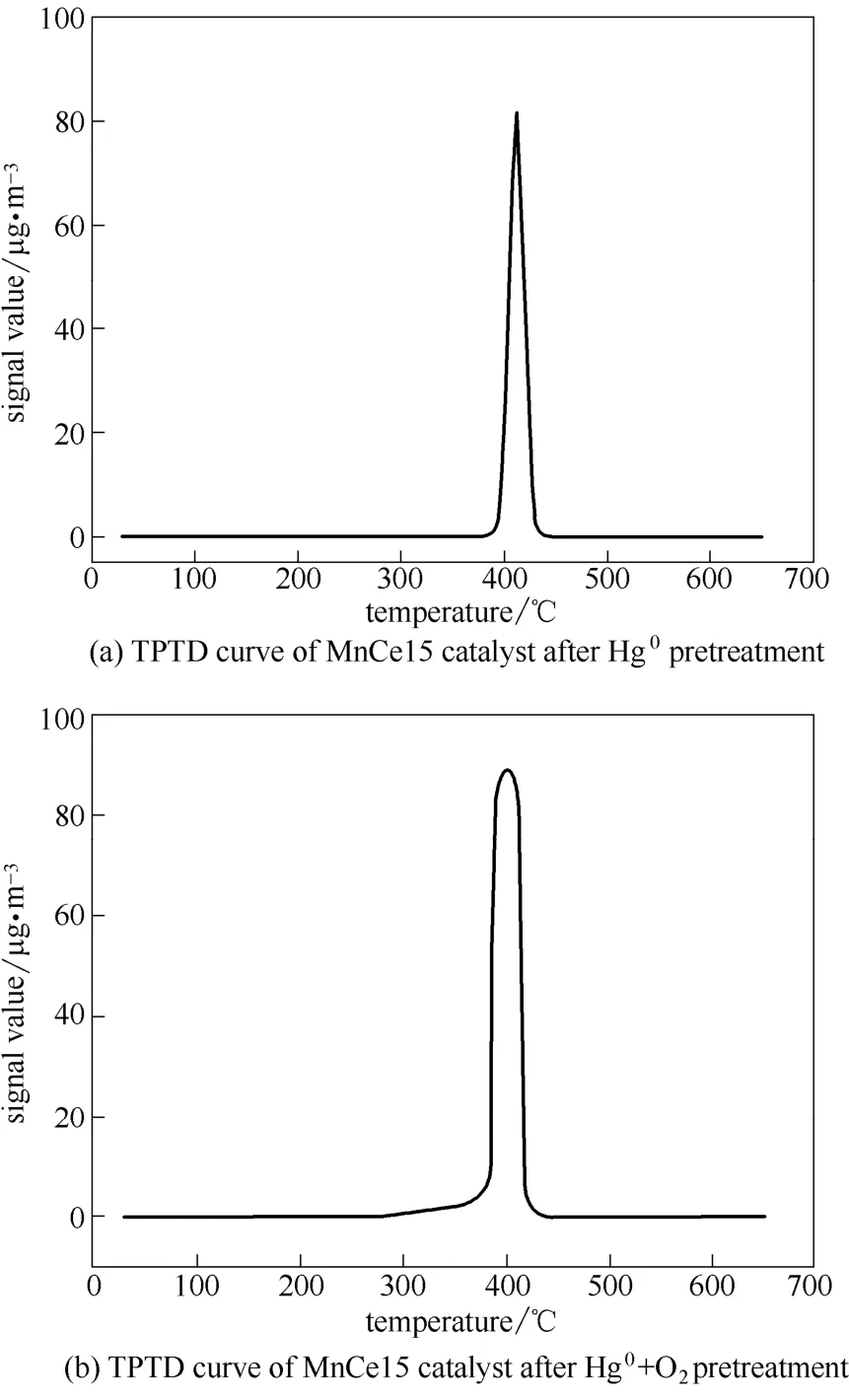
图4 预处理后的C1、C2 催化剂的TPTD 曲线Fig.4 TPTD curve of C1/C2 catalyst after pretreatme nt
C4 催化剂脱附峰的起止温度分别为60℃和150℃,峰值位于约100℃处。这一现象说明催化剂表面存在的二价汞为 HgCl2。基于 Langmuir- Hinshelwood 机理,HCl 会吸附在MnCe15 催化剂表面,并在有氧条件下被活化形成活性Cl,进而与吸附态的Hg0发生反应,生成HgCl2。图5(b)的结果证实了HgCl2的存在,为Langmuir-Hinshelwood机理提供了实验依据。
汞化合物在C5 催化剂上的程序升温热分解曲线如图6所示。C5 催化剂在450~550℃出现了一个分解脱附峰,峰值位于约400℃处。结合图3的结果可以发现C5 催化剂上主要存在的是HgSO4。SO2单独存在时,会与Hg0在MnCe15 催化剂上产生竞争吸附,但是在有氧的模拟烟气气氛下,即使SO2的浓度高达0.12%,催化剂仍然可以达到较高的Hg0氧化效率。可能的反应过程如下
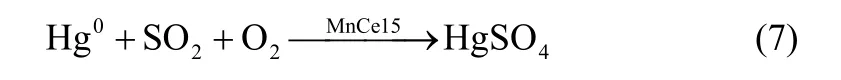

图5 预处理后的C3、C4 催化剂的TPTD 曲线Fig.5 TPTD curve of C3/C4 catalyst after pretreatment
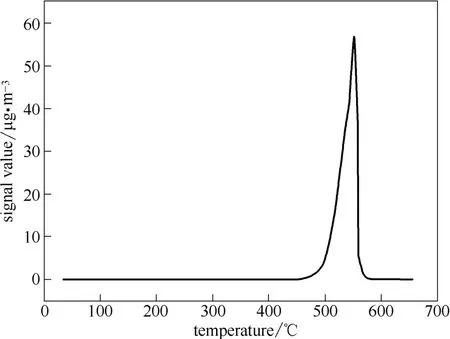
图6 预处理后的C5 催化剂的TPTD 曲线Fig.6 TPTD curve of MnCe15 catalyst after Hg0+SO2+O2 pretreatment
图7为C6 和C7 催化剂的TPTD 曲线。由图可以看出,在有氧和无氧条件下使用NO 处理过的 MnCe15 催化剂只能观察到HgO 存在,Hg(NO3)2并未在反应中生成,即NO 单独存在时并不能明显提高催化剂对Hg0的催化氧化活性。当通入O2后,虽然催化剂表面会形成多种含氮的活性物质,但是Hg0更倾向于与这些产生的活性物质反应生成HgO而非Hg(NO3)2。可能的反应路径如下


图7 预处理后的C6、C7 催化剂的TPTD 曲线Fig.7 TPTD curve of C6/C7 catalyst after pretreatment
对于在模拟烟气全气氛条件下预处理的C8 催化剂,为了防止不同的Hg0脱附峰的重叠,本节对程序升温过程进行了调整,依旧以10℃·min-1的升温速率将温度由室温升至650℃,但是在可能出现脱附峰的温度段将温度等温控制10~15 min,使得相应的汞化合物完全分解。具体的过程如下:第1 步,在80℃处停留,保证Hg2Cl2的分解;第2 步,在100℃处停留,保证HgCl2的分解;第3 步,在405℃处停留,保证Hg0的分解;第4 步,在533℃处停留,保证HgSO4的分解。
图8展示了C8 催化剂的程序升温热分解Hg0脱附曲线。汞化合物的分解发生在80~530℃范围内。在约110℃处有1 个明显的尖锐峰,表明HgCl2是C8 催化剂上主要存在的汞的物质形态,即在模拟烟气条件下MnCe15 催化剂对Hg0的催化氧化主要是将其氧化成为HgCl2。在450℃和530℃处的副峰表明HgO 和HgSO4可能出现在催化剂表面,但是浓度相对较低。在图8中并没有观察到Hg2Cl2的分解脱附峰。这一结果表明在烟气中HCl 是最主要的活性成分,对Hg0的催化氧化存在明显的促进作 用。煤种中氯的含量越高,SCR 催化剂对Hg0的氧化活性越好,结合后续的除尘和湿法脱硫装置可以高效地脱除燃煤烟气中的汞。
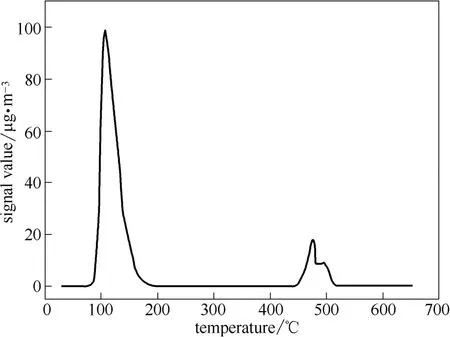
图8 预处理后的C8 催化剂的TPTD 曲线Fig.8 TPTD curve of MnCe15 catalyst after SFG pretreatment
3 结 论
为研究Hg0在MnCe15 催化剂上被催化氧化后在催化剂表面的赋存形态,并且为MnCe15 催化剂上Hg0的催化氧化反应机理提供更加充足的实验依据,使用程序升温热分解脱附手段对不同条件下预处理后的催化剂的Hg0脱附曲线进行了研究。得到的主要结论如下。
(1)证实了O2的存在可以补充反应中被消耗的晶格氧或化学吸附态氧,从而促进催化氧化反应发生。
(2)分析了HCl、SO2和NO 对Hg0在MnCe15表面发生催化氧化的反应方程式。在模拟烟气全气氛反应条件下,MnCe15 催化剂上Hg0的主要异相催化氧化产物是HgCl2,并且可以在催化剂表面检测到少量HgO 和HgSO4的生成。
(3)证实了汞与HCl 在MnCe15 催化剂上的氧化主要遵循Langmuir-Hinshelwood 机理,即HCl首先吸附在催化剂表面,被活化成为活性Cl,进而与邻近吸附位上的Hg0发生反应,生成HgCl2。
[1]Zhang Anchao (张安超),Xiang Jun (向军),Sun Lushi (孙路石),Hu Song (胡松),Fu Peng (付鹏),Cheng Wei (程伟),Qiu Jianrong (邱建荣).Synthesis and characterization of novel modified sorbents and their performance for elemental mercury removal [J].CⅠESC Journal(化工学报),2009,60 (6):1546-1553.
[2]Wang P,Su S,Xiang J,You H,Cao F,Sun L,Hu S,Zhang Y.Catalytic oxidation of Hg0by MnOx-CeO2/γ-Al2O3catalyst at low temperatures [J].Chemosphere,2014,101:49-54.
[3]He S,Zhou J,Zhu Y,Luo Z,Ni M,Cen K.Mercury oxidation over a vanadia-based selective catalytic reduction catalyst [J].Energy & Fuels,2008,23 (1):253-259.
[4]Eom Y,Jeon S,Ngo T,Kim J,Lee T.Heterogeneous mercury reaction on a selective catalytic reduction (SCR) catalyst [J].Catalysis Letters,2008,121 (3/4):219-225.
[5]Biester H,Scholz C.Determination of mercury binding forms in contaminated soils:mercury pyrolysisversussequential extractions [J].Environmental Science & Technology,1996,31 (1):233-239.
[6]Feng X,Lu J,Grègoire D,Hao Y,Banic C,Schroeder W.Analysis of inorganic mercury species associated with airborne particulate matter/aerosols:method development.[J].Analytical & Bioanalytical Chemistry,2004,380 (4):683-689.
[7]Bradshaw P,Koksoy M,Turkey W,Proceedings of the 23rd International Geology Congress [C].Academia,Prague,Section 7,1968:341-355
[8]Windmöller C C,Wilken R D,Jardim W D F.Mercury speciation in contaminated soils by thermal release analysis [J].Water,Air,and Soil Pollution,1996,89 (3/4):399-416.
[9]Biester H,Gosar M,Covelli S.Mercury speciation in sediments affected by dumped mining residues in the drainage area of the Idrija mercury mine,Slovenia [J].Environmental Science & Technology,2000,34 (16):3330-3336.
[10]Raposo C,Windmöller C C,DurãoJúnior W A.Mercury speciation in fluorescent lamps by thermal release analysis [J].Waste Management,2003,23 (10):879-886.
[11]Ozaki M,Uddin M A,Sasaoka E,Wu S.Temperature programmed decomposition desorption of the mercury species over spent iron-based sorbents for mercury removal from coal derived fuel gas [J].Fuel,2008,87 (17/18):3610-3615.
[12]Uddin M A,Ozaki M,Sasaoka E,Wu S.Temperature-programmed decomposition desorption of mercury species over activated carbon sorbents for mercury removal from coal-derived fuel gas [J].Energy & Fuels,2009,23 (10):4710-4716.
[13]Murakami A,Uddin M A,Ochiai R,Sasaoka E,Wu S.Study of the mercury sorption mechanism on activated carbon in coal combustion flue gas by the temperature-programmed decomposition desorption technique [J].Energy & Fuels,2010,24 (8):4241-4249.
[14]Wu S,Uddin M A,Nagano S,Ozaki M,Sasaoka E.Fundamental study on decomposition characteristics of mercury compounds over solid powder by temperature-programmed decomposition desorption mass spectrometry [J].Energy & Fuels,2010,25 (1):144-153.
[15]Rallo M,Lopez-Anton M A,Perry R,Maroto-Valer M M.Mercury speciation in gypsums produced from flue gas desulfurization by temperature programmed decomposition [J].Fuel,2010,89 (8):2157-2159.
[16]Lopez-Anton M A,Yuan Y,Perry R,Maroto-Valer M M.Analysis of mercury species present during coal combustion by thermal desorption [J].Fuel,2010,89 (3):629-634.
[17]Rallo M,Lopez-Anton M A,Meij R,Perry R,Maroto-Valer M M.Study of mercury in by-products from a Dutch co-combustion power station [J].Journal of Hazardous Materials,2010,174 (1/2/3):28-33.
[18]Lopez-Anton M A,Perry R,Abad-Valle P,Díaz-Somoano M,Martínez-Tarazona M R,Maroto-Valer M M.Speciation of mercury in fly ashes by temperature programmed decomposition [J].FuelProcessing Technology,2011,92 (3):707-711.
[19]Abad-Valle P,Lopez-Anton M A,Diaz-Somoano M,Martinez-Tarazona M R.The role of unburned carbon concentrates from fly ashes in the oxidation and retention of mercury [J].Chemical Engineering Journal,2011,174 (1):86-92.
[20]Rallo M,Heidel B,Brechtel K,Maroto-Valer M M.Effect of SCR operation variables on mercury speciation [J].Chemical Engineering Journal,2012,198/199:87-94.
[21]Rumayor M,Diaz-Somoano M,Lopez-Anton M A,Martinez-Tarazona M R.Mercury compounds characterization by thermal desorption [J].Talanta,2013,114:318-322.
[22]Presto A A,Granite E J.Survey of catalysts for oxidation of mercury in flue gas [J].Environmental Science & Technology,2006,40 (18):5601-5609.
[23]Niksa S,Fujiwara N.A predictive mechanism for mercury oxidation on selective catalytic reduction catalysts under coal-derived flue gas [J].Journal of the Air & Waste Management Association:Air & Waste Management Association,2005,55 (12):1866-1875.
猜你喜欢
化工管理(2022年13期)2022-12-02
中国新技术新产品(2022年6期)2022-07-03
温州大学学报(自然科学版)(2022年2期)2022-05-30
陶瓷学报(2020年3期)2020-10-27
测控技术(2018年2期)2018-12-09
制导与引信(2017年3期)2017-11-02
工业设计(2016年11期)2016-04-16
中国资源综合利用(2016年2期)2016-01-22
电网与清洁能源(2015年2期)2015-02-28
应用化工(2014年11期)2014-08-16
