
一种新型水合肼荧光比率探针的合成及应用研究
2015-07-17 00:22周秋兰谷标尹鹏李海涛
湖南师范大学学报·自然科学版 2015年3期
周秋兰 谷标 尹鹏 李海涛

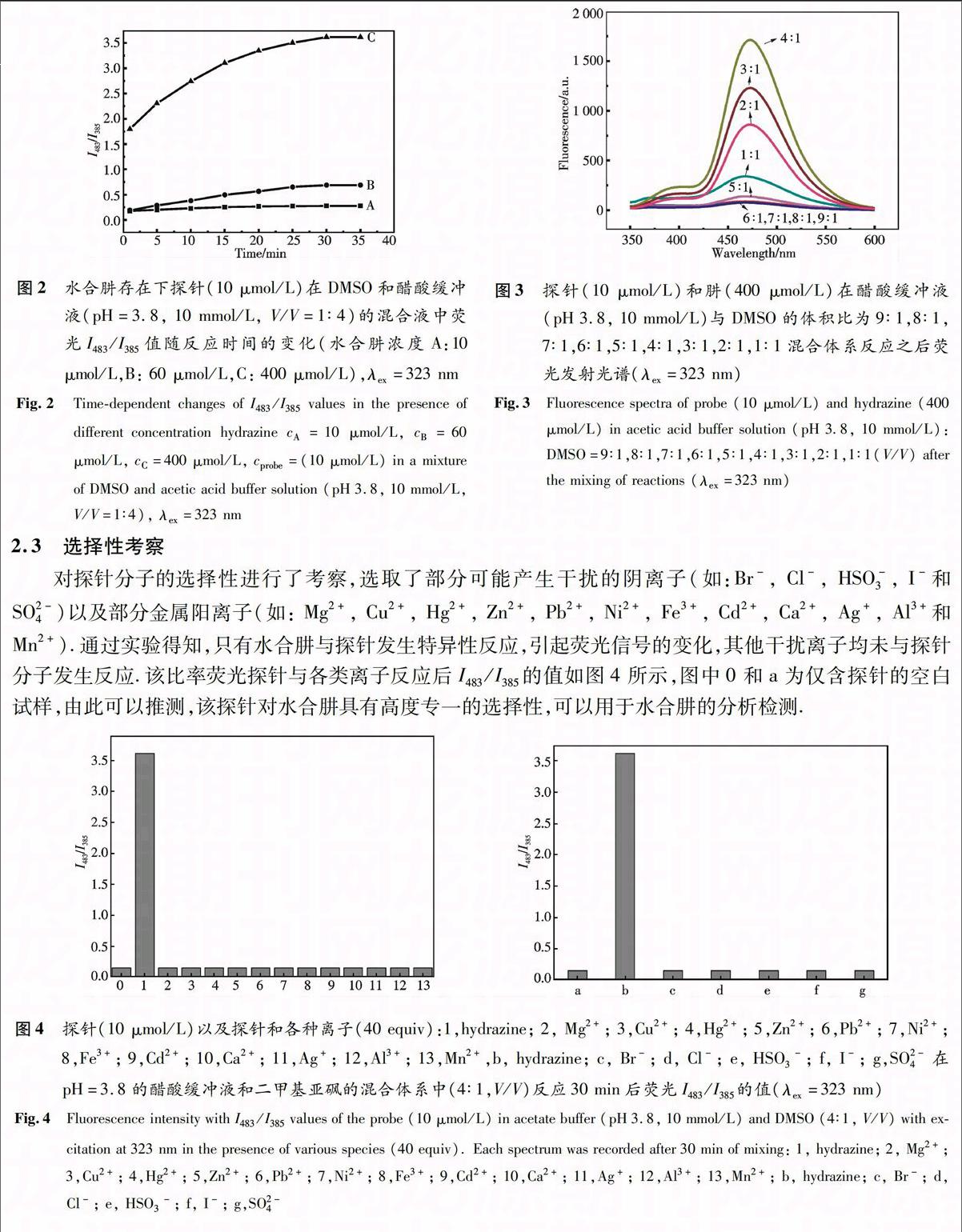
摘 要 基于激发态分子内质子转移的过程,设计合成了一种新颖的水合肼比率荧光探针—2-(苯并[d]噻唑-2-基)苯基-4-氧代戊酸甲酯(HBT-RCO),通过质谱(MS)和核磁共振(NMR)进行了结构表征.荧光光谱研究表明:在水合肼存在下,探针HBT-RCO的荧光光谱显著红移,且显示出比率荧光光谱特征,最大荧光强度比值(I483/ I385)增强约14倍.利用I483/ I385与水合肼浓度的比例关系实现对水合肼的定量检测.
关键词 比率荧光探针;2-(苯并[d]噻唑-2-基)苯基-4-氧代戊酸甲酯;水合肼;激发态分子内质子转移
中图分类号 O657.3 文献标识码 A 文章编号 1000-2537(2015)03-0047-06
肼(N2H4)常用作火箭推进剂、电池燃料以及有机合成[1-4]. 然而,肼易渗透皮肤组织或经口腔进入人体,破坏组织器官和神经系统[5-6].美国环保署规定肼的阈值为10 ppb[7]. 我国国家环境标准规定地面水和渔业水中肼的最高允许质量浓度10.0 μg/L[8].
肼的检测方法很多,如:气相色谱法、高效液相色谱法和电化学检测等[9-12],但是,这些方法设备昂贵,操作繁琐.荧光光谱分析因其具有高效、快速、灵敏度高等特点深受关注[13-19].如Choi[20]等设计出一种以香豆素为荧光团的基于光诱导电子转移(PET)的off-on型荧光探针用于肼的检测.但大多荧光探针都是单一的荧光增强或者猝灭,很难消除背景干扰.
比率荧光分析是利用荧光探针本身的发射波长处的荧光强度和与待测物反应后在另一发射波长处的荧光强度的比值作为检测信号来检测物质的一种方法[21].由于比率荧光探针以两个波长处荧光强度的比值作为信号参量,可以减少因探针浓度、设备以及样品环境等因素不可避免引起的误差,进而提高了选择性、灵敏度[22-23],在一定程度上解决了传统采集单一波长信号的探针分子存在的缺陷.
基于激发态分子内质子转移机理(ESIPT),我们开发设计出一种水合肼荧光探针,该探针中2-(2-羟基苯基)苯并噻唑(HBT)的羟基被乙酰丙酸保护起来,加入水合肼之后,与之发生环化和消除反应,探针荧光发生变化,最大吸收峰红移,而且紫外激发下呈现黄绿色荧光,从而可以实现对水合肼的比率荧光检测.
1 实验部分
1.1 仪器与试剂
试剂:2-羟基苯甲醛和2-氨基硫酚均购于上海德默医药科技有限公司,氨基磺酸和乙酰丙酸购于均购于安耐吉化学试剂公司,二氯亚砜、无水硫酸钠、乙腈和乙醇均购于国药集团化学试剂公司,均为分析纯试剂.丙酮和二氯甲烷均进行绝对无水处理,分析实验中均用二次蒸馏水,乙酸乙酯和石油醚均为常压蒸馏产物,硅胶(50~75 μm)经烘干处理.
仪器:PL303电子天平(梅特勒-托利多上海有限公司),ZNCL-G磁力搅拌器(巩义市予华仪器有限公司),DZF-6050真空干燥箱(上海飞越实验仪器有限公司),N-1001旋转蒸发仪(上海爱朗仪器有限公司),Bruker AVB-500核磁共振仪(瑞士Bruker公司),TU-1221紫外分光光度计(北京普析通用仪器有限公司),F-7000荧光分光光度计(日本Hitachi公司),PHS-3C 数字酸度计(上海鹏顺科学仪器有限公司).
1.2 探针的合成与表征
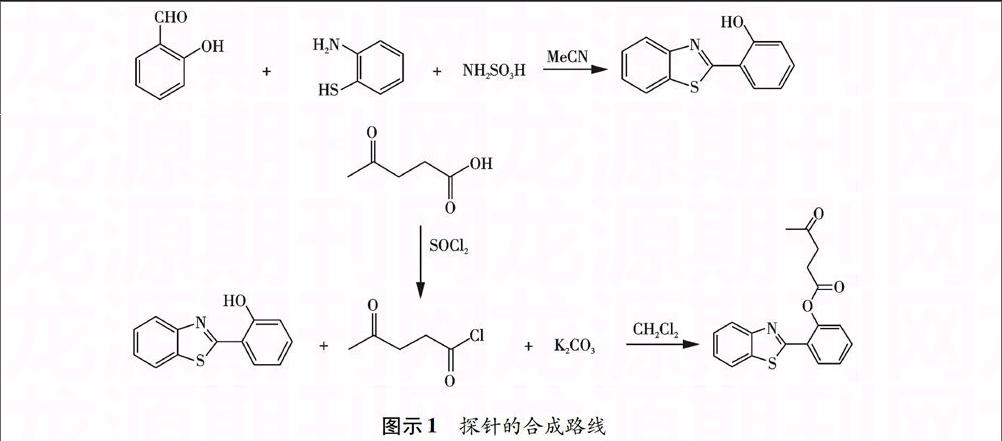
本文探针设计合成按照图示1所示路线,主要分为两步:第一步通过加成消去合成2-(2-羟基苯基)苯并噻唑(HBT);第二步通过取代反应将新制的乙酰丙酰氯与HBT反应,得到目标分子.
1.3 2-(2-羟基苯基)苯并噻唑(HBT)的合成
将2-羟基苯甲醛 (1.17 g ,9.58 mmol) 和2-氨基苯硫酚 (1.20 g, 9.58 mmol) 溶解在乙腈中 (20 mL),加入氨基磺酸作为催化剂(0.074 g, 0.074 mmol),在室温下搅拌反应3 h.反应过程用薄层色谱点板监控,得到固体物质,减压抽滤,然后水洗,最后将粗产品用乙醇重结晶,得到纯品 (1.85 g, 85.0%产率).1H NMR (500 MHz,CDCl3-d6),δ 12.52(1 H,s),7.96(1 H,d,J=1.0 Hz),7.89(1 H,d,J=1.0 Hz),7.69(1 H,d,J=1.0 Hz),7.48(1 H,d,J=1.0 Hz),7.39(2 H,m,J=3.0 Hz),7.10(1 H,t,J=1.0 Hz),6.95(1 H,d,J=1.5 Hz).
1.4 探针2-(苯并[d]噻唑-2-基)苯基-4-氧代戊酸甲酯的合成
先将乙酰丙酸 (464.5 mg, 4.00 mmol) 溶解在 SOCl2 (2 mL)中,在 N2保护下回流4 h. 用油泵抽干未反应完全的SOCl2 ,得到的油状物在冰浴下溶解在丙酮中(10 mL),加入合成得到的2-(2-羟基苯基)苯并噻唑 (227.3 mg, 1.00 mmol) 和碳酸钾 (552.8 mg, 4.00 mmol),在 N2 保护下室温搅拌,反应混合物倒入 20 mL CH2Cl2 ,然后用水洗3次. 收集有机相用无水硫酸钠干燥,旋转蒸发除去二氯甲烷,得到的粗产品用石油醚和乙酸乙酯混合液硅胶柱过柱分离,得到纯品 (246.1 mg ,75.6%产率) .1H NMR (500 MHz,CDCl3-d6),δ 8.19(1 H,d,J=1.0 Hz),7.95(1 H,d,J=1.0 Hz),7.76(1 H,d,J=1.0 Hz),7.36(2 H,t,J=2.5 Hz),7.24(2 H,dd,J=2.5 Hz),7.13(1 H,d,J=1 Hz)2.90(2 H,t,J=1.5 Hz),2.76 (2 H,t,J=1 Hz)2.06(3 H,s);13C NMR(500 MHz, CDCl3-d6), δ:206.19,171.07,162.36,152.86,148.12,135.23,131.31,130.08,126.30,126.21,125.87,125.25,123.63, 123.21,121.26,37.74,29.71, 28.61; MS(ESI+)[M+H]+:326.
猜你喜欢
当代化工(2020年2期)2020-03-18
中学课程辅导·教学研究(2017年29期)2018-02-26
分析化学(2017年12期)2017-12-25
金融理财(2015年7期)2015-07-15
海外星云 (2014年21期)2015-01-14
卷宗(2014年7期)2014-08-27
卷宗(2014年1期)2014-03-20
数理化学习·高一二版(2009年1期)2009-03-19
中学生物学(2008年12期)2008-12-27
中国科技术语(2004年4期)2004-03-18
