
钙同位素化学分离方法研究
2015-07-02 05:27:30刘峪菲祝红丽王桂琴许继峰张兆峰
地球化学 2015年5期
刘峪菲, 祝红丽, 刘 芳, 王桂琴, 许继峰*, 张兆峰*
(1. 中国科学院 广州地球化学研究所 同位素地球化学国家重点实验室, 广东 广州 510640; 2. 中国科学院大学, 北京 100049)
0 引 言
Ca作为主要的造岩元素及生物必需元素, 在许多地球化学及生物过程中起到重要作用, 因此对 Ca地球化学行为的研究能有效帮助我们认识多种地质过程。由于钙同位素之间存在的较大相对质量差(最大可达 20%), 除非选用最新的热电离质谱, 在一般常用仪器上很难实现Ca各个同位素的同时测定。此外, 钙同位素之间的丰度差异较大(40Ca/42Ca约为 151.016)[1–4],在测定过程中为了保证低丰度同位素及其比值的准确测定及获得较好的精度,40Ca信号需控制在15 V以上,这对仪器的要求较高, 且有可能损伤法拉第杯。但随着近年来质谱分析技术的发展, 克服这些困难准确测定钙同位素已成为可能, 这也为使用钙同位素示踪如洋壳物质再循环[5]、矿床形成[6]、古气候变化[7–8]、古海水演化[8–11]等地质过程提供了可能。
地质样品化学成分复杂, 因此在测试前必须经过离子交换柱的化学分离, 尽可能对Ca进行提纯以减少质谱测定过程中基体效应及同质异位素的干扰,从而获得高精度的钙同位素组成。值得注意的是Ca在离子交换柱上存在明显的分馏, 早在 1978年Russellet al.[4,12]发现由于树脂对各钙同位素的吸附程度不同导致 Ca在化学分离过程中发生同位素分馏, 分馏程度达 3‰, 接近 Ca在自然界的分馏。为了防止 Ca在离子交换柱上的分馏对测试造成影响,一般选择在样品中加入钙双稀释剂来校正柱子上的分馏。但综合考虑钙在离子交换柱上及质谱测定过程中的分馏效应, 在加入双稀释剂的情况下仍然要保证Ca的高回收率。因此, 无论在化学分离过程中是否加入双稀释剂, 都必须在保证基体效应较小的同时实现实验样品Ca的高回收率, 只有这样才能确保分离提纯的 Ca组分真实反映样品的钙同位素组成。我们针对影响化学分离效果的各种因素进行多组条件实验, 最终确定了一套Ca的淋洗曲线, 并以此为基础建立了Ca的化学分离流程。相较于前人标定的淋洗曲线[13–14], 我们选用 AG MP-50(100-200目)树脂及更低浓度的HCl淋洗, 在保证高回收率的情况下更有效地实现Ca与K、Sr的分离。
除化学分离影响样品真实的钙同位素组成外,空白也是一个重要因素。空白一般包括来自于实验器皿、试剂的空白及流程空白等, 在实验过程的每一个环节都可能对样品钙同位素造成影响。因此我们也对整个实验流程的空白进行了评估。
1 实验部分
1.1 试剂及器皿
实验使用的H2O均由Millipore公司的水纯化系统纯化, 为 18.2 MΩ。使用的 HCl、HNO3采用优级纯酸经亚沸蒸馏系统二次纯化制备, 使用的 HF则采用 BV-Ⅲ级酸纯化制备。实验所用的离子交换柱包括Bio-rad Poly-Prep聚丙烯层析柱(0.8 cm ID ×4 cm高度, 10 mL容量), 1 mL巴氏滴管自制的LDPE柱(0.5 cm ID × 13.5 cm高度, 3.5 mL容量)及Savillex PFA微柱(0.64 cm ID × 9 cm高度, 30 mL容量), 使用的其他器皿均为Teflon材质。在使用前所有聚丙烯及LDPE材质的离子交换柱均经过50% HNO3及50%HCl浸泡, 所有Teflon离子交换柱及器皿均经过HCl及HNO3反复煮沸清洗。实验使用的树脂为Bio-rad公司生产的AG MP-50(100-200目)及AG 50W-X8(200-400目)阳离子交换树脂, 两种树脂的官能团均为R-SO3-, 前者为具有物理孔的大孔型 AG 50W 树脂,对大分子物质具有较强的吸附及交换; 后者具有较高的交联度, 对离子具有较强的选择性。所有树脂在使用前必须经过6 mol/L HCl和8 mol/L HNO3反复浸泡。
1.2 仪器与标准
本次实验所有化学分离流程及仪器测试均在中国科学院广州地球化学研究所同位素地球化学国家重点实验室完成, 淋洗曲线的标定由ICP-OES仪器完成, 样品钙同位素测试经由Trion-TIMS仪器完成。
ICP-OES仪器以稀释 2000、4000、8000倍的BHVO-2标准溶液作为标准来确定各元素浓度并在测定过程中对仪器信号漂移进行监控。Trion-TIMS测试中选用42Ca-43Ca作为双稀释剂并依据指数分馏法则进行迭代计算以校正质量歧视效应。钙同位素组成采用通行的δ44/40Ca表示, 其表达式为δ44/40Ca (‰) =[(44Ca/40Ca)样品/(44Ca/40Ca)标样- 1]×1000。标样选用美国国家标准技术研究所(NIST)认证的NIST SRM 915a。
1.3 样 品
本文选用的样品包括碳酸盐标样、岩石标样、岩石样品及标准溶液。
碳酸盐标样: 国家标准GBW04412纯碳酸钙标准物质、NIST SRM 915a碳酸钙标准物质。
岩石标样: AGV-2、QLO-1、BIR-1a、BCR-2 4种美国国家地质局 (USGS)标准物质。岩石样品包括 T0049玢岩、T0031云英岩、T1377玄武岩、CM-10-04-12超钾质岩。
标准溶液: IAPSO大西洋标准海水及MISA多元素混合标准溶液。
为便于观察 Ca与 Sr的分离情况, 所有标定淋洗曲线的样品依据原有 Ca、Sr含量加入一定量 Sr标准溶液使样品中Ca/Sr = 2。
1.4 样品溶解
碳酸盐与硅酸盐样品采用不同的溶解流程。对于硅酸盐样品, 称取样品于7 mL杯子中, 将HF与HNO3按照 3∶1的比例加入样品, 摇匀后在 80 ℃电热板上保温一周后开盖蒸干, 再加入稀 HCl溶样并在 100 ℃电热板上加热 2 h, 蒸干后重复步骤直至样品完全溶解。碳酸盐则在称取样品后直接加入稀 HCl溶样, 蒸干后重复步骤至样品完全溶解。最后所有样品用稀HCl按照Ca浓度为1000×10–6进行定容, 用于化学分离。
2 结果及讨论
影响 Ca化学分离效果的因素主要包括离子交换柱类型、树脂体积、淋洗酸浓度及类型、上样量、样品岩性等, 我们针对各影响因素逐一设计条件实验, 通过标定Ca的最佳淋洗曲线确定了一套具有较好分离效果及高回收率的Ca化学分离流程。
2.1 离子交换柱的影响
在化学分离过程中, 离子交换柱的内径大小、长度都可能影响样品流经树脂的速率进而影响各元素的分离情况, 为了确定离子交换柱的影响, 我们选用Bio-rad Poly-Prep聚丙烯层析柱(0.8 cm ID × 4 cm高度, 10 mL容量)及Savillex PFA微柱(6.4 mm ID ×9 cm高度, 30 mL容量)两种柱子进行对比。
分别取 2 mL AG MP-50(100-200目)树脂装入Bio-rad Poly-Prep小柱及Savillex PFA微柱中, 清洗平衡柱子后取出含60 μg Ca的T0049玢岩样品上柱进行化学分离。样品上柱后依次使用60 mL 1.6 mol/L HCl、78 mL 2.5 mol/L HCl、8 mL 6 mol/L HCl淋洗。淋洗下来的溶液每2 mL接取1次, 由ICP-OES测试得到对应的淋洗曲线(图1)。由于两种离子交换柱的分离效果没有明显差别, 从柱子清洗及控制流程空白方面考虑, 我们优先选用Savillex PFA微柱。
此外, 从图中看出, 用1.6 mol/L HCl在约40 mL时已淋洗出来Ca, 换成2.5 mol/L HCl后由于酸浓度变化导致 Ca的峰形发生明显变化。两种离子交换柱的淋洗曲线中Al、Fe、K、Mg、Na、Ti都先Ca淋洗下来, 淋洗峰没有与Ca峰重合, 而后淋洗下来的Sr均有部分与Ca峰重合。在最后8 mL 6 mol/L HCl淋洗体积内没有出现明显淋洗峰表明在两种柱子上8种元素都已完全淋洗下来。另外对比用不同浓度HCl淋洗的Ca峰形, 2.5 mol/L HCl淋洗的峰形明显变陡, 峰区间也更窄, 表明淋洗的酸浓度越高Ca能越快淋洗下来。
2.2 树脂体积的影响
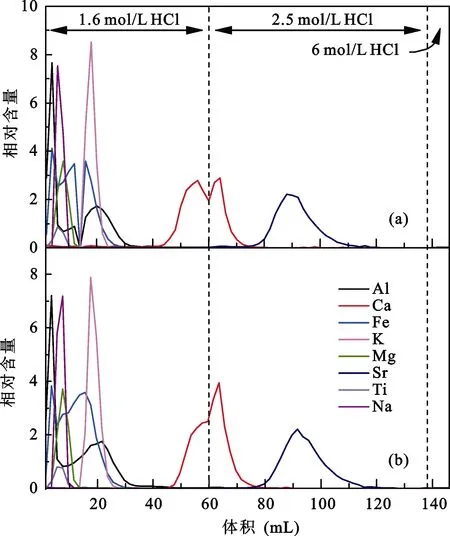
图1 不同离子交换柱的淋洗曲线Fig.1 Calcium elution curves on different chromatographic columns
化学分离使用的树脂类型及体积同样影响分离效果, 我们选用AG MP-50(100-200目)树脂分离Ca,并通过对比1 mL及2 mL树脂的淋洗曲线确定树脂体积的影响。取1 mL及2 mL AG MP-50(100-200目)树脂分别装入2根Bio-rad小柱中, 清洗及平衡柱子后, 取含60 μg Ca 的T0037云英岩样品上柱进行化学分离。为防止出现因酸浓度变化引起的Ca峰形变化, 调整淋洗酸浓度及体积。装有1 mL树脂的柱子依次使用22 mL 1.6 mol/L HCl、36 mL 1.8 mol/L HCl、4 mL 6 mol/L HCl淋洗, 淋洗溶液每1 mL接取1次。装有2 mL树脂的柱子依次使用44 mL 1.6 mol/L HCl、136 mL 1.8 mol /L HCl、4 mL 6 mol /L HCl淋洗, 淋洗溶液每2 mL接取1次。
对比两种树脂体积的淋洗曲线(图2a,图2b), Al、Fe、K、Mg、Na、Ti均能与Ca完全分离, 但Sr与Ca峰形都有重叠。2 mL树脂的淋洗曲线中(图2b)Ca峰形出现明显拖尾, 与Sr重叠部分比1 mL树脂中的范围宽, 并在最后4 mL 6 mol/L HCl出现小的淋洗峰, 表明之前淋洗的酸没有把Ca完全淋洗下来。此外, 2 mL树脂的淋洗体积约为1 mL树脂的2倍,导致实验流程时间加长将近1倍。因此, 使用2 mL树脂尽管能实现Ca与Al的有效分离, 但Ca峰形存在明显拖尾, 与Sr的分离效果不佳, 样品中Ca没有完全淋洗下来影响Ca回收率, 并且2 mL树脂使用酸的体积为1 mL树脂酸体积的两倍, 耗时也约为2倍,综合考虑我们最终确定使用 1 mL AG MP-50(100-200目)树脂。50 (100-200目)树脂淋洗曲线。为方便观察Sr淋洗曲线将Sr相对含量放大10倍
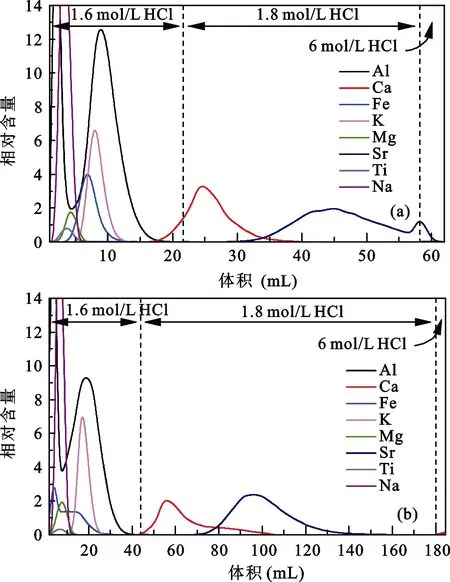
图2 不同树脂体积的淋洗曲线Fig.2 Calcium elution curves of different volumes of AG MP-50(100-200 mesh) cation exchange resin
(a) Calcium elution curves of 1mL AG MP-50 (100-200 mesh)cation exchange resin; (b) Calcium elution curves of 2 mL AG MP-50 (100- 200 mesh) cation exchange resin. Sr intensities were multiplied by 10 times for better display effects
2.3 淋洗酸的影响
2.3.1 不同酸浓度的影响
淋洗酸浓度的变化能明显影响化学分离中各元素的峰形及淋洗范围, 为确定酸浓度的影响程度,设置5组浓度连续变化的酸淋洗样品进行比较。选用 5根装有 1 mL AG MP-50(100-200目)树脂的Savillex微柱, 分别将含50 μg Ca的T0031云英岩样品上柱, 淋洗了22 mL 1.6 mol/L HCl后再分别用3 mol/L HCl、2.5 mol/L HCl、2.2 mol/L HCl、2.0 mol/L HCl、1.8 mol/L HCl淋洗35 mL, 最后用6 mol/L HCl淋洗5 mL, 淋洗溶液每1 mL接取1次。5组淋洗曲线(图3)中Ca淋洗峰均从17 mL开始出现, 能实现与Al、Fe、K、Mg、Na、Ti的分离。最后6 mol/L HCl淋洗部分都没有出现Ca淋洗峰, 表明样品中的Ca完全淋洗下来。随着淋洗酸浓度的降低, Ca峰形由陡变缓逐渐表现出拖尾现象, Ca的淋洗范围也随之变宽。Sr的淋洗峰随酸浓度降低而推迟出现, 峰形也随之变缓, 使Ca与Sr峰形重叠部分逐渐减小, 但也导致用浓度低于2 mol/L酸淋洗时Sr不能完全淋洗下来。
此外, 在前期使用2 mL AG MP-50(100-200目)树脂及Bio-rad小柱时曾用1.0 mol/L HCl淋洗T0031云英岩样品(图 3), 发现此时 Ca淋洗峰出现很晚,整个流程中 Ca峰没有完全出现, 柱子上仍残留有Ca, 因此要完全淋洗Ca需要提高酸浓度。基于这6组淋洗曲线的变化趋势, 我们尝试用 50 mL 1.6 mol/L HCl和5 mL 6 mol/L HCl淋洗样品并标定出淋洗曲线(图3)。1.6 mol/L HCl淋洗条件下Ca峰形更缓, 更明显的拖尾现象导致 Ca淋洗区间继续变宽, 但 Sr也更晚出现, 使与Ca的重叠部分仍然减小。因此我们最终确定Ca的化学分离流程中只采用1.6 mol/L HCl淋洗, 结合Ca峰形前后端与Al、Sr的重合情况,选定Ca的接取区间为18~44 mL, 共接取27 mL。
2.3.2 不同酸类型的影响
淋洗酸类型的不同也可能影响 Ca化学分离的效果, 为此选用浓度连续变化的 HNO3作为淋洗酸进行对比实验。由于AG 50W-X8(200-400目)树脂与AG MP-50(100-200目)树脂分离Ca的效果相近, 因此使用AG 50W-X8(200-400目)树脂标定Ca淋洗曲线不会产生明显变化。向4根装有1 mL AG 50W-X8(200-400目)树脂的 LDPE柱子加入含 50 μg Ca的CM-10-04-12超钾质岩样品, 考虑到用高浓度淋洗酸各元素分离效果较差, 分别选用60 mL 1 mol/L、1.1 mol/L、1.2 mol/L、1.3 mol/L HNO3淋洗, 每 1 mL接取1次, 标定出淋洗曲线(图4)。尽管使用HNO3淋洗样品, Ca与其他干扰元素能有很好的分离效果,但Ca淋洗峰出现明显晚于用HCl淋洗的, 且HNO3浓度变化对峰位置的影响明显大于 HCl, 综合考虑,我们仍选用HCl作为Ca化学分离的淋洗酸。
2.4 上样量及样品岩性的影响
除去实验试剂及器皿的影响, 样品的上样量及岩性也可能影响化学分离效果。为此我们设置一组含有 50 μg Ca的 GBW04412、100 μg Ca 的 MISA、50 μg Ca及100 μg Ca的T1377玄武岩样品的实验,其中GBW04412及MISA多元素混合标准溶液代表碳酸盐样品, T1377玄武岩代表硅酸盐样品。上样后用1.6 mol/L HCl淋洗65 mL, 每淋洗1 mL接取1次。从上样量看, 100 μg Ca的样品明显较50 μg Ca样品优先出现Ca淋洗峰且峰形更陡更窄。从岩性看碳酸盐样品的Ca峰都比硅酸盐样品的提前出现。但4种样品的 Ca峰区间均出现在设定的接取范围内,因此可以确定样品上样量在50~100 μg Ca对Ca的化学分离影响不大(图5a)。
由于不同岩性的样品Ca淋洗曲线存在变化, 设定的 Ca接取区间并不一定适用各种岩性。为确定Ca接取区间是否合适, 选用上样量为100 μg Ca的AGV-2、 QLO-1、BIR-1a、BCR-2、GBW04412 及T1377玄武岩6种样品进行化学分离。6种岩性样品Ca峰有所差别但基本上都出现在设定的接取范围内, 超出部分不到样品Ca总量的1%可忽略不计(图5b)。6种岩性样品在 Ca接取范围内均能保证 99%以上的回收率, 表明设定的接取区间仍能适用于大部分岩性样品, 样品岩性对Ca化学分离的影响在允许范围内。
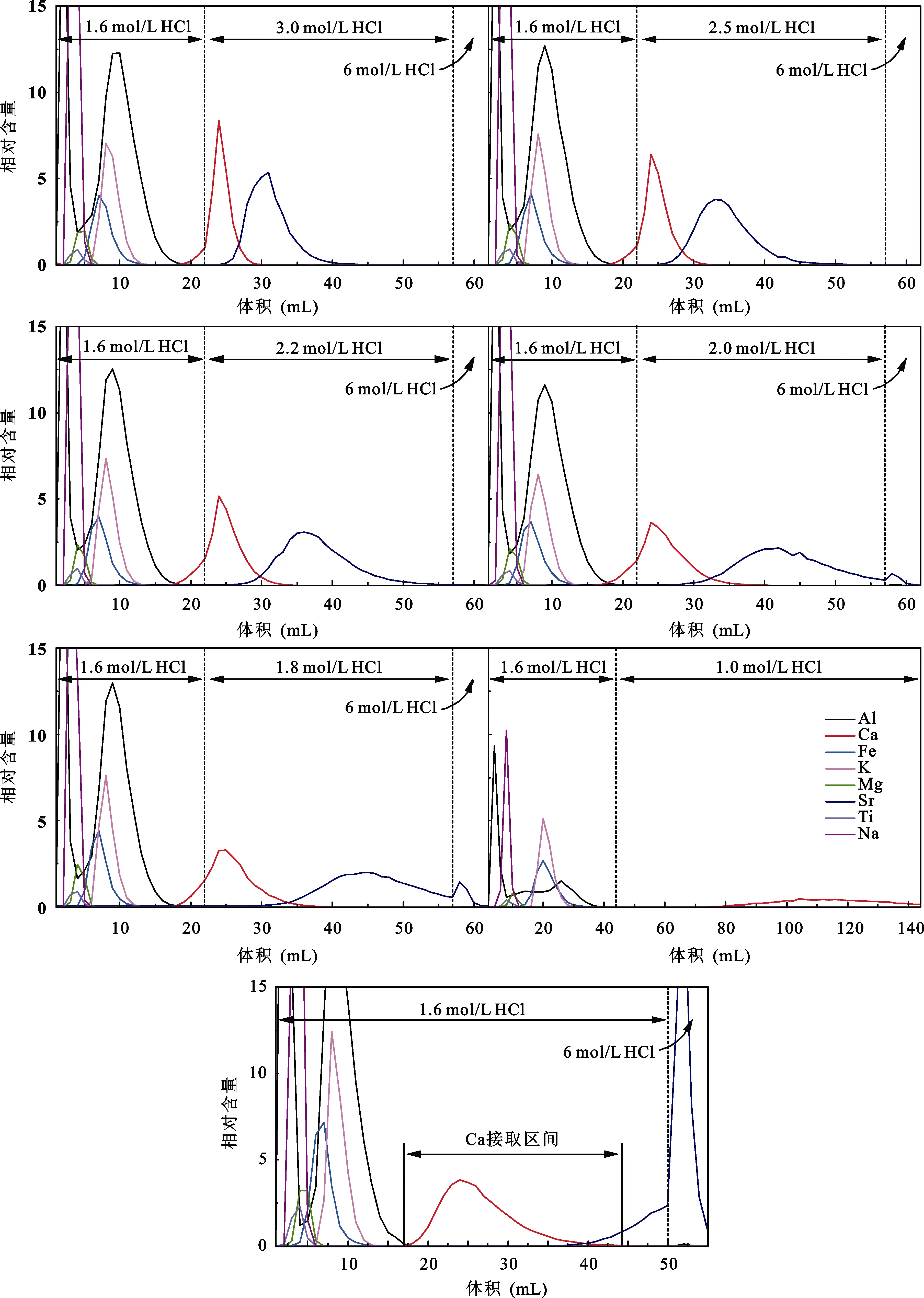
图3 不同HCl浓度的淋洗曲线Fig.3 Calcium elution curves by using different concentrations of HCl acid
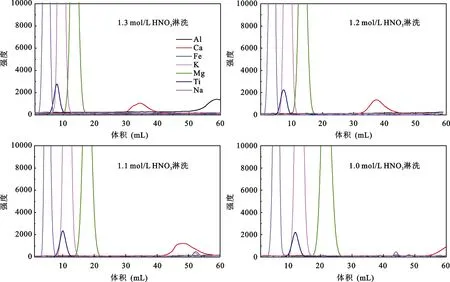
图4 不同HNO3浓度的淋洗曲线Fig.4 Calcium elution curves by using different concentrations of HNO3 acid
2.5 空白的影响
Ca属主量元素, 在化学分离过程中带入空白的量及组成也是影响样品 Ca分离效果的重要因素之一。空白主要来源于使用的试剂、器皿, 实验室环境及实验操作, 除保证试剂器皿洁净, 控制实验室本底及规范实验操作以减少带入空白外, 仍需要从溶样过程起带上至少1个重复样及空白来检测空白对样品的影响。使空白与样品经历完全相同的实验步骤, 通过TIMS测试加入了Ca双稀释剂的空白钙同位素组成, 计算全流程中空白的Ca总量以确定每次化学流程中空白的干扰。经过对多次流程空白的测定, 最终确定 Ca全流程空白为 20~100 ng, 已达国际其他实验室空白水平(表1)。此外, 还通过TIMS稀释剂法多次测试实验使用的 H2O、HCl、HNO3、HF及浸泡器皿的酸中的空白, 测得的空白均< 6 ng,不到全流程空白的 6%, 真正确保了实验试剂及器皿的洁净。依据50~100 μg Ca的上样量, 全流程空白占 Ca总量≤2‰, 对样品的影响可忽略不计。再者通过对比样品与重复样的测试结果, 发现两者δ44/40Ca值在误差范围一致, 进一步表明化学分离流程中空白对样品的影响不大, 流程空白达到实验要求。
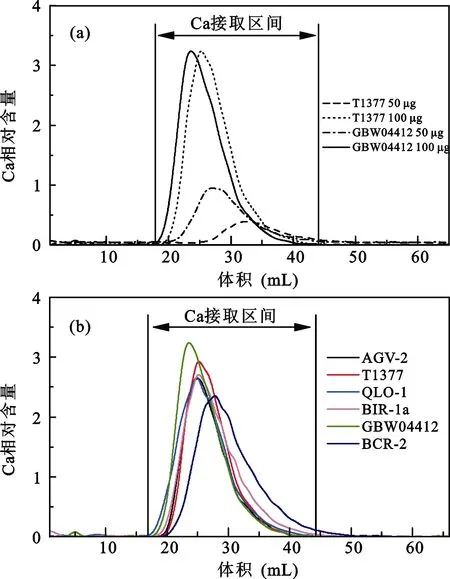
图5 Ca的接取区间Fig.5 Range of calcium cut
2.6 TIMS测试
结合各组条件实验结果最终标定了一套可行的淋洗曲线(图6c)。与目前已发表的淋洗曲线[12–13](图6a,图6b)相比, 标定的淋洗曲线中Ca与K、Sr的分离效果明显更好。在此基础上我们建立了一套具有高回收率且可更好分离Ca与K、Sr的化学分离流程。须注意的是对实际测定钙同位素组成的样品必须在化学分离前加入 Ca双稀释剂来校正离子交换柱上的分馏。另外在分离流程中除接取Ca组分, Ca前后的淋洗溶液也一并接取下来用ICP-OES检测Ca含量以监控回收率情况。
在实际测试样品前, 我们在 TIMS上检测样品中各基体元素如40K、48Ti、84Sr、86Sr、88Sr、24Mg19F、25Mg19F、24Mg16O、27Al16O 等信号强度, 确定基体元素的信号强度均低, 对 Ca信号影响可忽略不计,表明建立的 Ca化学分离流程能有效控制质谱测试过程中的基体效应。另外我们设置两组分别在过柱前加稀释剂和在过柱后加稀释剂的对比实验来检测化学分离流程中Ca的回收率情况(表2)。测试结果发现两组δ44/40Ca值均在误差范围一致, 且与文献值[2-9,15-18]一致, 说明离子交换柱上没有Ca残余, 化学分离流程中Ca实现高回收率。

表1 Ca全流程空白对比Table 1 Comparison of total procedural Ca blank in different laboratories
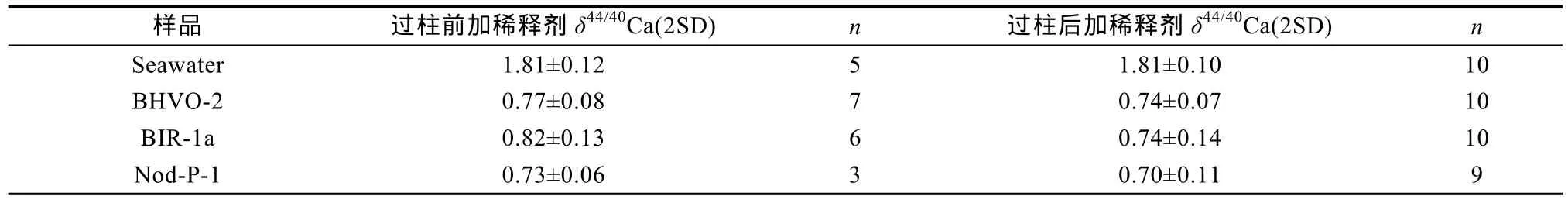
表2 化学分离前后加入稀释剂的结果对比Table 2 Comparison of δ44/40Ca values spiked before and after chemical separation
最后, 通过对经化学分离提纯的 NIST SRM915a及IAPSO大西洋标准海水2个标样的多次实测, 得到δ44/40Ca915a=0.01±0.11 (2SD)或±0.02(2SE),n=46;δ44/40Caseawater=1.81±0.10(2SD)或±0.02(2SE),n=19。标样的结果均与文献中的测试结果[2,9,15–18]在误差范围内一致, 表明建立的化学分离方法能有效去除干扰元素的影响, 真实反映出样品的钙同位素组成。
3 结 论
本文基于对离子交换柱类型、树脂体积、酸浓度及类型、上样量、样品岩性等影响化学分离效果因素的实验研究及对实验流程空白的评价, 建立了一套 Ca与 K、Sr分离效果较好、回收率高的钙同位素化学分离方法。方法采用装有1 mL AG MP-50 100-200目树脂的Savillex PFA微柱作为离子交换柱,以1.6 mol/L HCl淋洗上样量50~100 μg Ca的样品,在淋洗体积为18~44 mL时接取Ca。方法全流程空白控制在20~100 ng, 和国际同类实验室相当。经这套化学分离方法提纯的NIST SRM 915a、IAPSO大西洋标准海水及3种USGS标样测试结果与文献值在误差范围内一致, 表明建立的化学分离流程满足钙同位素高精度测定的要求。
本研究得到国家自然科学基金面上项目(41373007)、重大项目(41490632)、重点项目(91328204)及同位素地球化学国家重点实验室基金(SKLIG-JY-12-01,SKLIG-JY-13-03,
SKLIG-KF-13-03)的资助; 实验过程中得到康晋霆同学以及马金龙、孙胜玲、刘颖等老师的支持与帮助; 在文章审稿过程中得到两位审稿人的中肯建议及编辑的辛勤帮助,在此一并致谢!
:
[1] Boulyga S F. Calcium isotope analysis by mass spectrometry[J]. Mass Spectrom Rev, 2010, 29(5): 685–716.
[2] Depaolo D J. Calcium isotopic variations produced by biological, kinetic, radiogenic and nucleosynthetic processes[J].Rev Mineral Geochem, 2004, 55(1): 255–288.
[3] 李亮, 蒋少涌. 钙同位素地球化学研究进展[J]. 中国地质,2008, 35(6): 1088–1100.Li Liang, Jiang Shao-yong. Advance in calcium isotope geochemistry[J]. Geol China, 2008, 35(6): 1088–1100 (in Chinese with English abstract).
[4] Russell W A, Papanastassiou D A, Tombrello T A. Ca isotope fractionation on the Earth and other solar system materials[J].Geochim Cosmochim Acta, 1978, 42(8): 1075–1090.
[5] Huang S, Farka J, Jacobsen S B. Stable Calcium isotopic compositions of Hawaiian shield lavas: Evidence for recycling of ancient marine Carbonates into the mantle[J].Geochim Cosmochim Acta, 2011, 75(17): 4987–4997.
[6] Zhu Hong-li, Ling Ming-xing, Zhang Zhao-feng, Sun Wei-dong. Calcium isotopic constraints on the genesis of the Bayan Obo deposit[C]. 2013.
[7] Christina L, Depaolo D J. Isotopic evidence for variations in the marine Calcium cycle over the Cenozoic[J]. Science, 2000,289(5482): 1176–1178.
[8] Griffith E M, Paytan A, Caldeira K, Bullen T D, Thomas E. A dynamic marine Calcium cycle during the past 28 million years[J]. Science, 2008, 322(5908): 1671–1674.
[9] Farkaš J, Böhm F, Wallmann K, Blenkinsop J, Eisenhauer A,Van Geldern R, Munnecke A, Voigt S, Veizer J. Calcium isotope record of Phanerozoic oceans: Implications for chemical evolution of seawater and its Causative mechanisms[J]. Geochim Cosmochim Acta, 2007, 71(21): 5117–5134.
[10] 柯婷, 韦刚健, 刘颖, 谢露华, 邓文峰, 王桂琴, 许继峰.南海北部珊瑚高分辨率硼同位素组成及其对珊瑚礁海水pH变化的指示意义[J]. 地球化学, 2015, 44(1): 1–8.Ke Ting, Wei Gang-jian, Liu Ying, Xie Lu-Hua, Deng Wen-feng, Wang Gui-qin, Xu Ji-feng. High resolution boron isotopic compositions of a coral from the northern South China Sea and their implications for reconstruction of seawater pH[J]. Geochimica, 2015, 44(1): 1–8 (in Chinese with English abstract).
[11] Farkaš J, Buhl D, Blenkinsop J, Veizer J. Evolution of the oceanic Calcium cycle during the late Mesozoic: Evidence fromδ44/40Ca of marine skeletal Carbonates[J]. Earth Planet Sci Lett, 2007, 253(1): 96–111.
[12] Russell W A, Papanastassiou D A. Calcium isotope fractionation in ion-exchange chromatography[J]. Anal Chem,1978, 50(8): 1151–1154.
[13] Huang S C, Farkaš J, Jacobsen S B. Calcium isotopic fractionation between clinopyroxene and orthopyroxene from mantle peridotites[J].Earth Planet Sci Lett, 2010, 292(3): 337–344.
[14] Wieser M E, Buhl D, Bouman C, Schwieters J. High precision Calcium isotope ratio measurements using a magnetic sector multiple collector inductively coupled plasma mass spectrometer[J]. J Anal Atom Spectrom, 2004, 19(7): 844–851.
[15] Holmden C, Bélanger N. Ca isotope cycling in a forested ecosystem[J]. Geochim Cosmochim Acta, 2010, 74(3): 995–1015.
[16] Amini M, Eisenhauer A, Böhm F, Holmden C, Kreissig K, Hauff F, Jochum K P. Calcium Isotopes (δ44/40Ca) in MPI-DING reference glasses, USGS rock powders and various rocks:Evidence for Ca isotope fractionation in terrestrial silicates[J].Geostand Geoanal Res, 2009, 33(2): 231–247.
[17] Hippler D, Schmitt A D, Gussone N, Heuser A, Stille P,Eisenhauer A, Nägler T F. Calcium isotopic composition of various reference materials and seawater[J]. Geostand Newslett, 2003, 27(1): 13–19.
[18] Huang S, Farkaš J, Yu G, Petaev M I, Jacobsen S B. Calcium isotopic ratios and rare earth element abundances in refractory inclusions from the Allende CV3 chondrite[J]. Geochim Cosmochim Acta, 2012, 77: 252–265.
猜你喜欢
食品安全导刊(2023年30期)2023-11-10 07:31:26
中国化工贸易·上旬刊(2020年5期)2020-09-10 07:22:44
聚氯乙烯(2018年5期)2018-02-18 03:30:28
聚氯乙烯(2018年5期)2018-02-18 03:30:28
材料科学与工程学报(2016年2期)2017-01-15 13:34:42
电源技术(2015年7期)2015-08-22 08:49:04
化学分析计量(2015年5期)2015-03-23 20:36:21
同位素(2014年3期)2014-06-13 08:22:28
同位素(2014年2期)2014-04-16 04:57:15
同位素(2014年2期)2014-04-16 04:57:12
