
α-O-4型木质素二聚体热解行为的理论研究*
2015-06-23 16:24武书彬刘超邓裕斌
华南理工大学学报(自然科学版) 2015年6期
武书彬 刘超 邓裕斌
(华南理工大学制浆造纸工程国家重点实验室,广东广州510640)
α-O-4型木质素二聚体热解行为的理论研究*
武书彬 刘超 邓裕斌
(华南理工大学制浆造纸工程国家重点实验室,广东广州510640)
为揭示热解过程中木质素中α-O-4连接键的解聚机理,运用密度泛函理论模拟了α-O-4型木质素二聚体模化物苯酚基甘油-α-苯基醚在773K、101 kPa条件下的热解行为.通过对各步反应热力学焓变的计算,明确了二聚体的初次裂解是Cα—O键和Cα—Cβ键的断裂.根据热力学可能性,将二聚体的后续裂解设计成不同的路径,热解的最终产物有小分子化合物(甲醇、乙醇、乙二醇和环氧乙烷)、苯酚及其对位取代物(对甲基苯酚、对乙基苯酚和对羟基苯甲醇)、苯及其取代物(苯甲醇),酚类产物中较优先的是对甲基苯酚、对乙基苯酚,其次是苯酚.此外,还将α-O-4型二聚体的热解行为与β-O-4型二聚体进行了比较,发现其不同均源于两者结构的差异.
木质素二聚体;热解;密度泛函理论;α-O-4连接键
木质素是由苯基丙烷结构单元构成的、具有空间网状结构的天然大分子,是植物体的三大主要组分之一,对植物起到了支撑作用.木质素的年产量很大,仅次于纤维素,且具有高热值,可以通过热解的方式将其转化为生物油[1-3],而热解是现阶段生物质资源化利用的主要手段之一.国内外关于木质素热解的研究较多,并系统地给出了木质素在不同工况条件下的热解特性和产物分布[4-7].
木质素的热解是一个十分复杂的过程,很多学者为解释其机理做了大量工作,他们通过实验或者计算机模拟建立动力学方程来解释热解的动力学过程[8-10].Britt等[11]提出热解是一个自由基的反应过程.现阶段对热解机理的研究则是通过模化物的模拟实验[12-13]和量化计算实现的.Huang等[14]对木质素单体模化物愈创木酚的热解路径进行了研究.Beste等[15-16]研究了含有不同取代基的木质素二聚体模化物苯乙基苯基醚(PPE)的键解离能和反应速率常数,而Huang等[17]给出了PPE可能的热解过程和热解产物.
木质素主要通过醚键(β-O-4、α-O-4、4-O-5)和碳碳键(β-5、5-5、β-1、β-β)连接[18],其中β-O-4连接键的含量最高,达45%~60%[19-23],所以在进行机理探究时,一般选择具有β-O-4连接键的模化物作为研究对象[24-26].但是,木质素中约有10%~15%的醚键是以α-O-4和4-O-5的形式存在的[19-23],它们和β-O-4一样,也具有相对较高的活性,也影响着木质素的热解过程和产物分布.然而,此类研究却鲜见报道.文中运用密度泛函理论对α-O-4型木质素二聚体的热解行为进行了过程模拟,预测了可能的热解路径和产物.
1 理论计算
1.1 计算模型
文中选取具有α-O-4特征连接键的木质素二聚体模化物苯酚基甘油-α-苯基醚(简记为PGE)作为研究对象,其结构如图1所示.
1.2 计算方法
文中所有的计算均在Gaussian 03软件包[27]中进行.首先用半经验方法PM3对木质素二聚体的各种可能构象进行优化,然后用B3LYP/6-31G(d)这一密度泛函方法作筛选,得到能量最低构象,并以此构象来进行后续的计算.文中出现的反应物、中间体和产物经过频率验证均无虚频,为能量极小值的稳定结构;过渡态经过频率验证均有唯一虚频,且通过了內禀反应坐标(IRC)方法的验证.优化结构后,用B3LYP/6-311G(d,p)进行了单点能的计算,条件为773K、101kPa.
文中讨论的是基于以下反应的焓变:
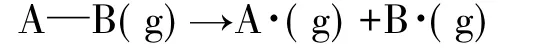
一定温度下的焓值(H(T))由以下表达式进行估算:
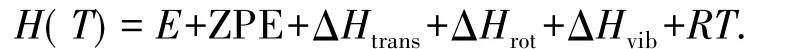
其中,A、B为不同的基团,T为反应温度,E是分子总能量,ZPE是零点能,ΔHtrans、ΔHrot、ΔHvib分别代表平动、转动和振动对能量的贡献,R是普适气体常量.
焓变表达式为
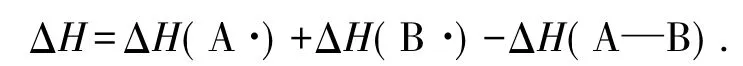
2 结果与讨论
2.1 二聚体的初次裂解
木质素及其模化物在热解时总是优先发生侧链C3结构的解聚,所以文中设计了如图2所示的PGE可能的初次裂解路径.其中,路径R1、R2、R3分别是C1—Cα、Cα—Cβ、Cβ—Cγ键的断裂,α-O-4键的断裂分为R4、R5两个路径,分别是芳基醚键和烷基醚键的断裂,Cβ上羟基的解离按路径R6进行,Cα和Cβ上氢原子的裂解则设计成路径R7和R8.
图2中给出了各步路径的热力学焓变(ΔH).从数值上可以看出,所有路径的焓变均大于零,即单从热力学角度来看,这些路径都是不能自发进行的,需要从外界吸收能量反应才能进行.其中,路径R4的焓变最小,其次是R2,分别为180.33和238.45 kJ/mol,其余各路径的热力学焓变都很大,分布在309.91~374.09kJ/mol之间.这表明路径R4和R2需要外界提供的能量最少,最易进行.也就是说,路径R4和R2在初次裂解时会优先进行,即 Cα—O醚键和Cα—Cβ键会优先裂解.
2.2 二聚体的后续裂解
上述分析给出了PGE热解的两条优先路径,下面分别对这两条路径的后续裂解过程进行探讨.由Cα—Cβ键断裂引发的裂解如图3所示,图4给出的则是由Cα—O键断裂引发的裂解.需要说明的是,各物质在第一次出现时都尽可能详尽地给出了其变化,再次出现时,只给出了热力学优先的产物.
2.2.1 Cα—Cβ键断裂引发的裂解
PGE按路径R2热解生成P1和P2,P1和P2后续可能的热解过程被设计成路径R2-1-R2-29,如图3所示.其各可能步骤的热力学焓变如表1所示.
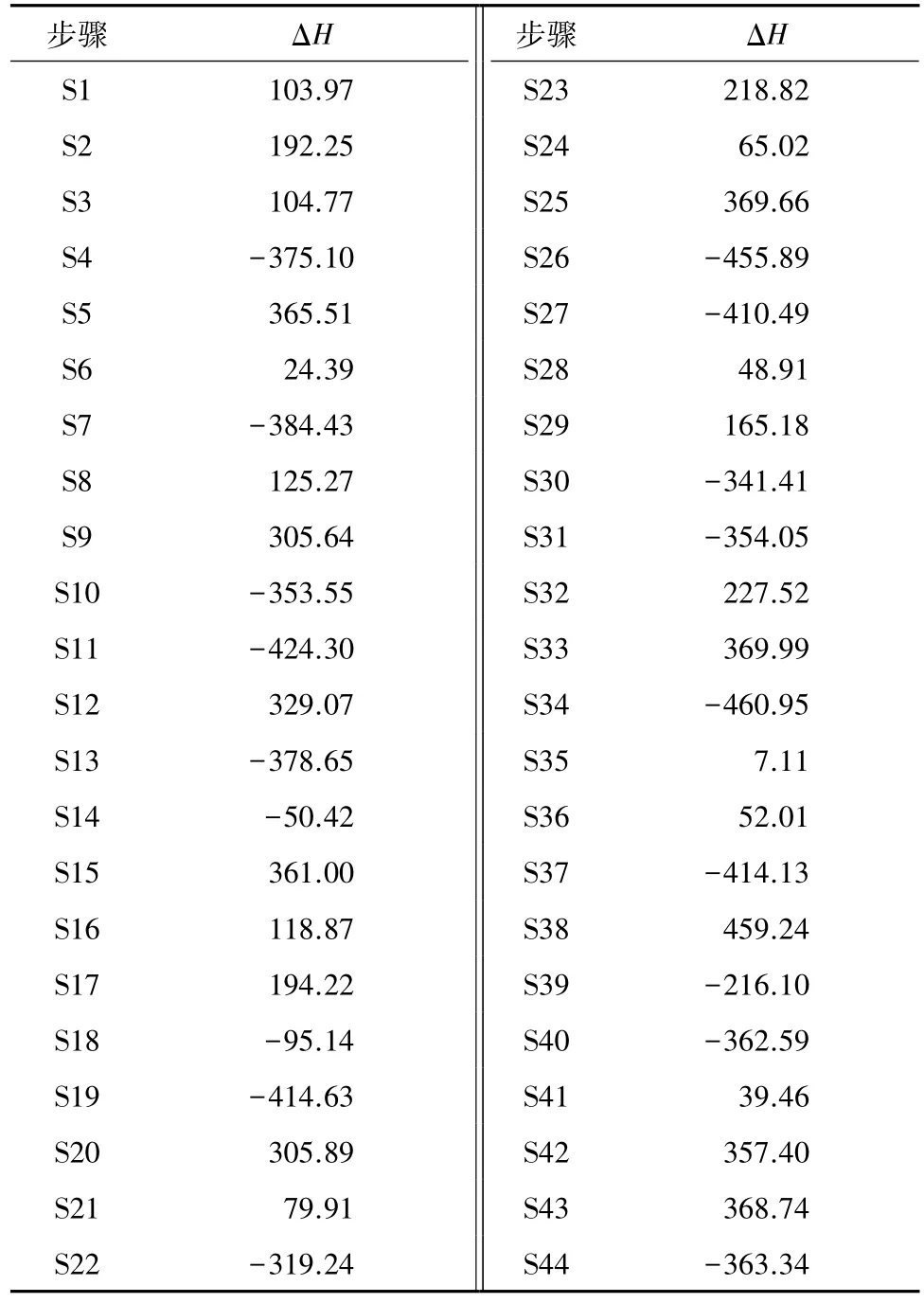
表1 Cα—Cβ键断裂引发的裂解过程中各可能步骤的焓变Table 1 Calculated enthalpy changes of all possible steps in the pyrolysis process of PGE caused by the dissociation of Cα—Cβbound kJ/mol
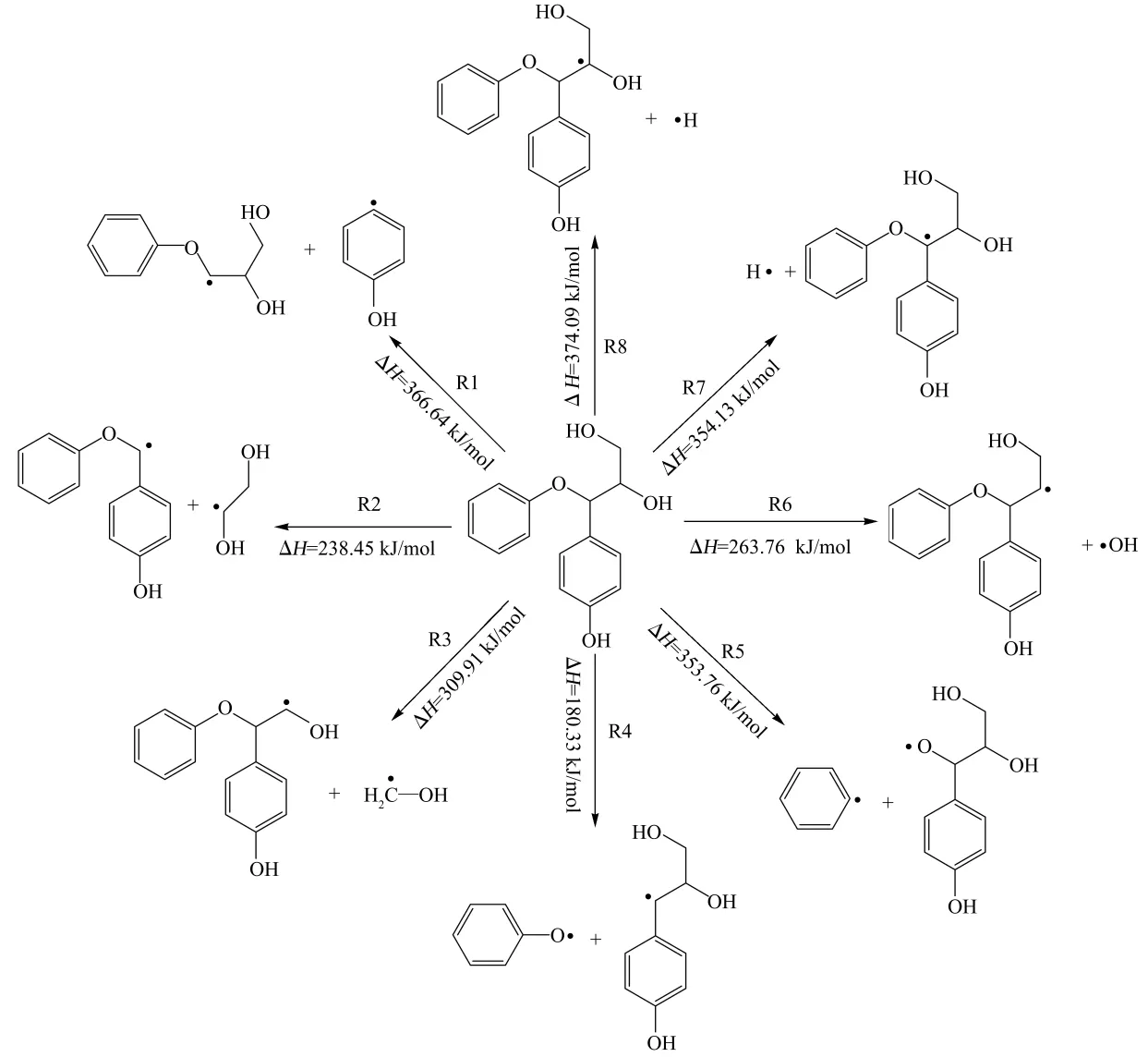
图2 PGE可能的初次裂解路径Fig.2 Possible first pyrolysis paths of PGE
首先,P1失氢生成P3,P3存在着3种可能的裂解方式,分别是羟基乙醛中醛基氢的解离、C—C键和C—O键的断裂.按S5生成的P7会发生脱羰反应,放出CO,同时生成P8,P8可能被氢自由基中和生成P9,或者脱氢生成P10,完成路径R2-1和R2-2,这两条路径的焓变分别为347.90和857.59kJ/mol,均大于零,热力学可能较小.按S9生成P8和P11,P8转变成热力学优先产物P9,P11则可能被氢自由基或羟基自由基中和成P10或P12,路径R2-3、R2-4和 R2-5的焓变分别为 263.63、294.51和223.76kJ/mol,也大于零,不是热力学优先的过程.按S12失去羟基自由基后生成P13,再夺氢生成P14,此过程R2-6的焓变为292.84 kJ/mol,大于零.由P3裂解而设计的6条路径的焓变都大于零,单从热力学角度来看,都不会优先发生.
接着,P1可能按S2的方式失氢,闭环生成P4,或者按S3的方式失去羟基自由基生成P5,由于烯醇式结构的不稳定性,P5会与P14发生结构互变.路径R2-7和R2-8的焓变分别为430.70和292.80 kJ/mol,需要外界提供能量,同样不是热力学可能的过程.
P1还能够与氢自由基结合生成P6,此过程放出375.10 kJ/mol的能量,使得从PGE开始生成P6的焓变为-136.65kJ/mol,小于零,说明P6(乙二醇)是热力学可能的产物.在热化学的作用下,P6还会继续裂解.C—O键按照S15断裂生成P15,P15分别失去羟基自由基、氢自由基或与氢自由基中和生成P5、P16、P17和P18,同样地,P5互变异构成P14,路径R2-9-R2-12的焓变分别为-190.29、129.20、418.57和292.80kJ/mol,显然,只有路径R2-9是热力学可能的,产物P18(乙醇)是可能的产物.C—C键按照S20断裂生成两分子P8,P8再转变成热力学优先产物P9,整个过程的焓变为-215.18kJ/mol,小于零,说明路径R2-13是热力学可能的过程,产物P9(甲醇)是可能的产物.P6还可能分子内脱水,闭环生成P16,这一过程的焓变为-56.74kJ/mol,小于零,说明路径R2-14是热力学可能的,产物P16(环氧乙烷)是可能的产物.

P19结构中两芳环之间的Ar—O键、C—O键和Ar—C键容易断裂,从而引起P19的再次裂解.首先,Ar—O键均裂,按照S25生成P22和P23,P22与氢结合成较为稳定的P24,P23则存在着两种可能的变化,分别生成P25和P26.路径R2-15、R2-16和R2-17的焓变分别为-121.63、337.77和-167.03kJ/mol,说明按R2-15和R2-17生成P25和P24是热力学可能的.其次,C—O键均裂,按照S29生成P27和P28,P27转变成稳定的P29,P28则发生夺氢或失氢反应,生成P30和P31.路径R2-18、R2-19和R2-20的焓变分别为-256.90、-269.66和311.92kJ/mol,说明按R2-18和R2-19生成P29和P30是热力学可能的.最后,Ar—C键均裂,生成P32和P33,同样地,P32与氢结合成稳定的P29,P33会结构重排生成P34,然后夺氢或失氢生成P35和P36.路径R2-21、R2-22和R2-23的焓变分别为-171.75、348.31和-117.82 kJ/mol,说明按R2-21和R2-23生成P29和P35是热力学可能的.纵观路径R2-15-R2-23,P19的3种断键方式都是热力学可能的,其会按照各自热力学优先的过程分别生成P24(苯)、P25(对羟基苯甲醇)、P29(苯酚)、P30(对甲基苯酚)和P35(苯甲醇).
P20的裂解只设计了一种方式,即按S38进行的Ar—O键的断裂.生成的P22转变成热力学优先的P24,生成的P37则两次夺氢,最终生成P26.路径R2-24和R2-25的焓变分别为337.82和460.62kJ/mol,是吸热反应,热力学的优先性较小.P21失氢转变成具有二苯甲酮稳定结构的P39只需外界提供很少的能量就可以实现,P39的裂解分别按S42和S43的方式断裂Ar—C键,化学键的均裂产物分别按照热力学优先的过程与氢中和生成P24、P26、P29和P36.路径R2-26-R2-29的焓变分别为244.43、337.73、250.71和348.32kJ/mol,大于零,均是吸热反应,都不是热力学优先的过程.
通过对Cα—Cβ键断裂引发PGE后续裂解的分析可以看出,P1会优先与氢结合生成 P6,再按R2-9、R2-13和R2-14分别生成乙醇、甲醇和环氧乙烷,且R2-13的反应活性要高于R2-9和R2-14.P2则会优先与氢结合生成P19,然后分别按3种方式断键,最终生成苯、对羟基苯甲醇、苯酚、对甲基苯酚和苯甲醇,各可能路径反应活性的相对大小为R2-19、R2-18>R2-21、R2-17>R2-15、R2-23.
2.2.2 Cα—O键断裂引发的裂解
PGE的热解由于Cα—O键断裂而生成P41和P27,P41和P27后续的热解过程如图4所示,分别按路径R4-1-R4-17进行.其各可能步骤的热力学焓变如表2所示.

表2 Cα—O键断裂引发的裂解过程中各可能步骤的焓变Table 2 Calculated enthalpy changes of all possible steps in the pyrolysis process of PGE caused by the dissociation of Cα—O bound kJ/mol
首先是P27的变化.2.2.1节的分析已经表明,P27会夺氢生成热力学的优先产物P29,即为路径R4-1.此路径的焓变为-161.08 kJ/mol,属放热反应,是热力学可能的过程,其可能产物是苯酚.
其次是从P41开始的后续变化.P41可能与氢中和生成P42;可能脱去羟基自由基形成双键,从而生成P43;或者发生电子的转移,形成P44.在P44中,由于自由基和羟基处在同一个碳上,不稳定,故P44会失氢转变成较为稳定的羰基化合物P53.下面接着探讨P42、P43和P53可能的变化.
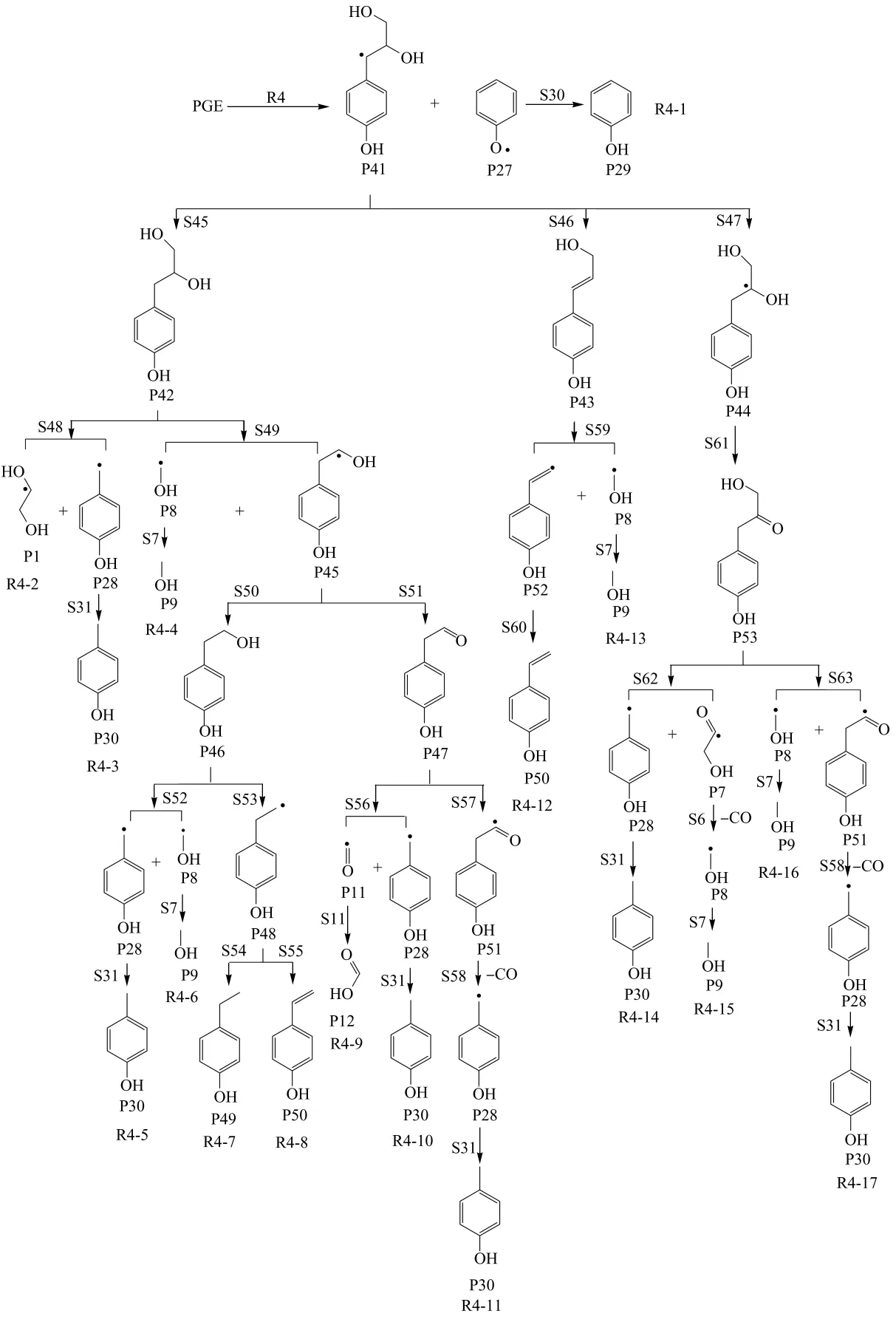
图4 Cα—O键断裂引发的裂解Fig.4 Pyrolysis of PGE caused by the dissociation of Cα—O bound
由于侧链结构中C—C键断裂位置的不同,P42的裂解有S48和S49两种方式.按S48生成P1和P28,P1最终转变成甲醇、乙醇、乙二醇或环氧乙烷等小分子物质,为路径R4-2.P28则转变成热力学优先的P30,为路径R4-3,此路径的焓变为-272.71kJ/ mol,是热力学可能的过程,可能的产物为对甲基苯酚.按S49生成的P8经由S7生成P9,路径R4-4的焓变为-256.56 kJ/mol,属于放热反应,此过程是热力学可能的过程,甲醇是可能的产物.按S49生成的P48由于存在夺氢与失氢两种可能性,会分别按S50和S51生成P46和P47.P46的裂解按C—C键和C—O键断键方式的不同分为S52和S53两步,生成的P8和P28按热力学优先性转变成P9和P30,完成路径R4-5和R4-6;生成的P48则有可能转变成P49和P50,完成路径R4-7和R4-8.这4条路径的焓变分别为-341.92、-372.29、-300.70和232.88kJ/mol,其中R4-5-R4-7是热力学可能的过程,可能的热力学产物为甲醇、对甲基苯酚和对乙基苯酚.P47的裂解则可能发生侧链C—C键的断裂,生成P11和P28,最终生成P12和P30;或者脱去醛基氢,生成P51,继而脱羰生成P28,P28又转变成热力学优先的P30.由P47引发的路径R4-9、R4-10和R4-11的焓变分别为66.69、136.94和221.21kJ/mol,均大于零,都不是热力学可能的过程.
P43的裂解只设计了一种断键方式,即按S59生成P8和P52,两者再与氢结合生成热力学优先的P9和P50,完成路径R4-12和R4-13.两条路径的焓变分别为232.84和296.44 kJ/mol,属吸热反应,热力学可能性较小.同样地,由于侧链结构中C—C键断裂位置的不同,P53的裂解有S62和S63两种方式,生成的P7和P28、P8和P51分别按一步或多步转变成热力学优先的产物,完成路径R4-14-R4-17.由P53引发的4条路径的焓变分别为196.82、190.83、205.64和221.21 kJ/mol,均大于零,都是吸热反应,单从热力学角度来看,都不是可能的反应过程.
从上述分析可以看出,由Cα—O键断裂引发的PGE的后续裂解中,路径R4-1-R4-7的焓变小于零,是热力学可能的过程,其反应活性的相对大小为R4-2、R4-5、R4-6>R4-7>R4-3、R4-4>R4-1,可能的产物为环氧乙烷、乙二醇、甲醇、苯酚、对甲基苯酚和对乙基苯酚.
纵观PGE热解的整个过程,热力学可能的热解路径有多个,可能的热解产物包括小分子(环氧乙烷、乙二醇、乙醇和甲醇)、苯酚及其对位取代物(对甲基苯酚、对乙基苯酚和对羟基苯甲醇)、苯及其取代物(苯甲醇),产物的可能性与真实木质素热解得到的生物油中的主要组分(酚类化合物)是一致的.其中,产生酚类化合物最优先的路径是R4-5和R4-7,最优先的酚类产物是对甲基苯酚和对乙基苯酚.
2.3 与β-O-4型二聚体热解的比较
通过分析具有类似结构、不同特征链接方式的两种模化物的热解行为,对比其主要链接键的断裂顺序、可能的反应路径和主要的反应产物,可以更好地掌握木质素的热解行为,了解某些产物的结构来源.因此,文中对比α-O-4型和β-O-4型[24]木质素二聚体的热解行为,发现两者具有几点相似之处.首先,在初次裂解时,总是侧链的烷基醚键最先断裂,然后是Cα—Cβ键的断裂,而其他链接键的断裂则相对较难.其次,可能的最终产物都是苯酚及其取代物,且都伴随有小分子生成.当然,两者的热解行为也存在着不同.首先,小分子物质的种类不同,α-O-4型热解生成乙二醇、环氧乙烷、乙醇和甲醇,而β-O-4型热解生成乙醇,并无其他种类的小分子物质.其次,两者的主要产物不同,α-O-4型热解的主要产物是对甲基苯酚和对乙基苯酚,而β-O-4型热解的主要产物是苯酚.最后,在β-O-4型的热解产物中出现了对位取代基中含有不饱和羰基的物质,如对羟基苯甲醛,而在α-O-4型的热解产物中没有出现.所有的这些不同都是由于两者结构的差异造成的,了解这些差异能够更好地把握天然木质素的热解行为.
3 结论
(1)运用密度泛函理论对木质素二聚体模化物苯酚基甘油-α-苯基醚的热解行为进行了模拟,通过各反应的热力学焓变给出了二聚体初次裂解最有可能的路径,即Cα—O和Cα—Cβ键断裂,这与β-O-4型二聚体热解时初次裂解的规律一致,总是侧链的烷基醚键最先断裂,然后是Cα—Cβ键断裂,而其他链接键的断裂则相对较难.由此可认为,木质素在热解时应该是结构中的醚键及Cα—Cβ键优先断裂.
(2)Cα—Cβ键断裂引发的后续热解中只有路径R2-9、R2-13-R2-15、R2-17-R2-19、R2-21和R2-23是热力学可能的,同样地,Cα—O键断裂引发的后续热解中只有R4-1-R4-7是热力学可能的.所有路径的热力学可能性依次为R4-6、R4-5>R4-7>R4-3、R2-19>R2-18、R4-4>R2-13、R2-9>R2-21、R2-17、R4-1>R2-15、R2-23>R2-14.最终可能的热力学产物有小分子物质(甲醇、乙醇、乙二醇和环氧乙烷)、苯酚及其对位取代物(对甲基苯酚、对乙基苯酚和对羟基苯甲醇)、苯及其取代物(苯甲醇),其中最优先产生的酚类化合物是对甲基苯酚、对乙基苯酚,其次是苯酚.
(3)α-O-4型二聚体热解的主要产物是小分子物质和酚类化合物,这与β-O-4型的热解产物在种类、结构和优先性上是不同的.两种模化物热解行为的对比表明,这些不同均源于两者结构的差异,从不同的特征产物可以反推出物质原本的结构,以此可以把握木质素的热解行为.
[1] Bridgwater A V,Peacocke G.Fast pyrolysis processes for biomass[J].Renewable and Sustainable Energy Reviews,2000,4:1-73.
[2] Bridgwater A V,Meier D,Radlein D.An overview of fast pyrolysis of biomass[J].Organic Geochemistry,1999,30 (12):1479-1493.
[3] Luo Z,Wang S,Liao Y,etal.Research on biomass fastpyrolysis for liquid fuel[J].Biomass and Bioenergy,2004,26(5):455-462.
[4] Caballero J A,Font R,Marcilla A.Pyrolysis of kraft lignin:yields and correlations[J].Journal of Analytical and Applied Pyrolysis,1997,39:161-183.
[5] Jiang G,Nowakowski D J,Bridgwater A V.Effect of the temperature on the composition of lignin pyrolysis products[J].Energy&Fuels,2010,24(8):4470-4475.
[6] Guo D L,Wu SB,Liu B,et al.Catalytic effects of NaOH and Na2CO3additives on alkali lignin pyrolysis and gasification[J].Applied Energy,2012,95:22-30.
[7] 娄瑞.非木材纤维木素在不同热化学条件下的产物形成规律与调控途径[D].广州:华南理工大学轻工与食品学院,2011.
[8] 武书彬,向冰莲,刘江燕,等.工业碱木素热裂解特性研究[J].北京林业大学学报,2008,30(5):143-147.
Wu Shu-bin,Xiang Bing-lian,Liu Jiang-yan,et al.Pyrolysis characteristics of technical alkali lignin[J].Journal of Beijing Forestry University,2008,30(5):143-147.
[9] Ferdous D,Dalai A K,Bej SK,et al.Pyrolysis of lignins: experimental and kinetics studies[J].Energy&Fuels,2002,16(6):1405-1412.
[10] Beste A,Buchanan Iii A C.Kinetic analysis of the phenyl-shift reaction inβ-O-4 lignin model compounds:a computational study[J].The Journal of Organic Chemistry,2011,76(7):2195-2203.
[11] Britt P F,Buchanan Iii A C,Thomas K B,et al.Pyrolysis mechanisms of lignin:surface-immobilized model compound investigation of acid-catalyzed and free-radical reaction pathways[J].Journal of Analytical and Applied Pyrolysis,1995,33:1-19.
[12] Kotake T,Kawamoto H,Saka S.Pyrolysis reactions of coniferyl alcoholas amodelof the primary structure formed during lignin pyrolysis[J].Journal of Analyticaland Applied Pyrolysis,2013,104:573-584.
[13] Kuroda K,Nakagawa-Izumi A.Analytical pyrolysis of lignin:products stemming fromβ-5 substructures[J].Organic Geochemistry,2006,37(6):665-673.
[14] Huang J,LiX,Wu D,etal.Theoretical studies on pyrolysis mechanism of guaiacol as lignin model compound[J].Journal of Renewable and Sustainable Energy,2013,5:043112.
[15] Beste A,Buchanan Iii A C.Computational study of bond dissociation enthalpies for lignin model compounds.Substituent effects in phenethyl phenyl ethers[J].The Journal of Organic Chemistry,2009,74(7):2837-2841.
[16] Beste A,Buchanan Iii A C.Substituent effects on the reaction rates of hydrogen abstraction in the pyrolysis of phenethyl phenyl ethers[J].Energy&Fuels,2010,24 (5):2857-2867.
[17] Huang X,Liu C,Huang J,et al.Theory studies on pyrolysismechanism of phenethyl phenyl ether[J].Computational and Theoretical Chemistry,2011,976(1):51-59.
[18] Kang S,Li X,Fan J,et al.Hydrothermal conversion of lignin:a review[J].Renewable and Sustainable Energy Reviews,2013,27:546-558.
[19] Adler E.Lignin chemistry-past,present and future[J].Wood Science and Technology,1977,11(3):169-218.
[20] Pandey M P,Kim C S.Lignin depolymerization and conversion:a review of thermochemicalmethods[J].Chem Eng Technol,2011,34(1):29-41.
[21] Chakar F S,Ragauskas A J.Review of current and future softwood kraft lignin process chemistry[J].Industrial Crops and Products,2004,20(2):131-141.
[22] Rodrigues Pinto P C,Borges Da Silva E A,Rodrigues A E.Insights into oxidative conversion of lignin to highadded-value phenolic aldehydes[J].Industrial&Engineering Chemistry Research,2010,50(2):741-748.
[23] Shen D K,Gu S,Luo K H,et al.The pyrolytic degradation of wood-derived lignin from pulping process[J].Bioresource Technology,2010,101(15):6136-6146.
[24] 王华静,赵岩,王晨,等.木质素二聚体模型物裂解历程的理论研究[J].化学学报,2009,67(9):893-900.Wang Hua-jing,Zhao Yan,Wang Chen,et al.Theoretical study on the pyrolysis process of lignin dimer model compounds[J].Acta Chimica Sinica,2009,67(9):893-900.
[25] Drage TC,Vane CH,AbbottGD.The closed system pyrolysis ofβ-O-4 lignin substructure model compounds[J].Organic Geochemistry,2002,33(12):1523-1531.
[26] Chu S,Subrahmanyam A V,Huber G W.The pyrolysis chemistry of aβ-O-4 type oligomeric lignin model compound[J].Green Chemistry,2013,15(1):125-136.
[27] Frisch M J,Trucks GW,Schlegel H B,etal.Gaussian 03[CP].Revision E.01.Wallingford C T:Gaussian Inc,2014.
Theoretical Investigation into Pyrolysis Behaviors ofα-O-4 Lignin Dimer
Wu Shu-bin Liu Chao Deng Yu-bin
(State Key Laboratory of Pulp and Paper Engineering,South China University of Technology,Guangzhou 510640,Guangdong,China)
In order to reveal the depolymerizationmechanism ofα-O-4 linkages in lignin during the process of pyrolysis,phenolglycerol-α-phenyl ether is selected as theα-O-4 lignin dimermold compound,and its pyrolysis behaviors at 773K and 101kPa are simulated according to the density functional theory(DFT).Then,the enthalpy changes of each step are calculated.It is found that the dimer pyrolysis starts from the dissociations of Cα—O and Cα—Cβbonds.According to the thermodynamics possibility,the following pyrolysis is designed into different paths,and the final pyrolytic products are found to contain smallmolecule compounds(methanol,ethanol,ethy-lene glycol and ethylene oxide),phenol and para-substituted phenols(p-methyl phenol,p-ethyl phenol and p-hydroxybenzyl alcohol),benzene and substituted benzene(benzyl alcohol).In all these phenolic compounds,p-methyl phenol and p-ethyl phenol are themost possible products,which are followed by phenol.In addition,the pyrolysis behaviors ofα-O-4 andβ-O-4 lignin dimers are compared.The results show that all the differences between the two lignin dimers are attributed to their different structures.
lignin dimer;pyrolysis;density functional theory;α-O-4 linkage
s:Supported by the National Program on Key Basic Research Project of China(2013CB228101)and the National Natural Science Foundation of China(31270635,21176095)
TK 6;O6-39
10.3969/j.issn.1000-565X.2015.06.004
1000-565X(2015)06-0022-08
2014-03-10
国家“973”计划项目(2013CB228101);国家自然科学基金资助项目(31270635,21176095)
武书彬(1965-),男,教授,博士生导师,主要从事植物纤维类生物质化学结构、植物纤维类生物质转化为清洁能源和化工原料等的研究.E-mail:shubinwu@scut.edu.cn
猜你喜欢
能源化工(2021年6期)2021-12-30
中学生数理化(高中版.高考理化)(2021年5期)2021-07-16
云南化工(2020年11期)2021-01-14
合成技术及应用(2021年1期)2021-01-07
上海包装(2019年8期)2019-11-11
天津造纸(2016年1期)2017-01-15
电子制作(2016年19期)2016-08-24
制冷技术(2016年4期)2016-08-21
中国造纸学报(2015年1期)2015-12-16
天津城建大学学报(2015年5期)2015-12-09
