
氟西汀对小鼠肝脏Ces1d,Ces1e和CYP3A11抑制作用及机制
2015-06-12 12:37王宇文储栋宝陈瑞妮
遵义医科大学学报 2015年1期
尚 伟,王宇文,储栋宝,熊 晶,刘 娓,刘 杰,陈瑞妮,杨 俭
(南京医科大学 药理学系,江苏 南京 210029)
基础医学研究
氟西汀对小鼠肝脏Ces1d,Ces1e和CYP3A11抑制作用及机制
尚 伟,王宇文,储栋宝,熊 晶,刘 娓,刘 杰,陈瑞妮,杨 俭
(南京医科大学 药理学系,江苏 南京 210029)
目的 探讨氟西汀对小鼠肝脏药物代谢酶(Ces1d,Ces1e和CYP3A11)的作用及机制。方法 雄性C57BL/6N小鼠分为对照组(CON)、氟西汀低剂量组、氟西汀低剂量组和LPS组,分别腹腔注射等容积生理盐水、氟西汀10、20 mg/kg和LPS 2 mg/kg,连续3 d。提取肝脏组织蛋白,测定酶活性,Western Blot分析药物代谢酶(Ces1d、Ces1e、CYP3A11)、PXR和Stra13的表达。结果 氟西汀明显降低小鼠肝脏总水解酶活性以及羧酸酯酶(Ces1 d,Ces1e)的表达(P<0.05、0.01);明显降低小鼠肝脏CYP3A11活性以及氧化酶(CYP3A11)的表达(P<0.05、0.01);氟西汀能浓度依赖性降低调节药物代谢酶的核受体PXR的表达(P<0.05、0.01),同时增加转录因子Stra13的表达(P<0.05、0.01)。结论 氟西汀降低药物代谢酶(Ces1d、Ces1e、CYP3A11)的表达及活性可能与其降低PXR表达和增加Stra13表达有关。
氟西汀;药物代谢酶;PXR;Stra13
肝脏含有多种在药物代谢中发挥重要作用的药物代谢酶[1],羧酸酯酶(Carboxylesterase, CES)属于B族酯酶,组成一个多基因家族,其基因产物广泛定位于机体多种组织和器官的内质网(ER)中,这些酶可以有效地催化水解一系列内源性和外源性物质,包括含酯键、酰胺键和硫酯键的药物[2-3]。肝脏主要表达羧酸酯酶1(Carboxylesterase 1,CES1,Ces1d,小鼠)和羧酸酯酶2(Carboxylesterase 2,CES2,Ces1e,小鼠)。细胞色素P4503A4(Cytochrome P450 3A4,CYP3A4)是肝脏中最重要、承担大约50%以上现存处方的药物代谢[4],药物代谢酶是产生药物相互作用主要因素,具有非常重要的药理学和毒理学意义[4-5]。
抑郁症是全球共同关注的公共卫生健康问题慢性精神疾患[6],预计至2020年将成为第二大全球疾病负担[7]。选择性5-羟色胺再摄取抑制剂(selective serotonin reuptake inhibitors, SSRIs)是治疗抑郁症的主要药物,其中以氟西汀(fluoxetine, FLX)应用最为广泛[8]。临床上在氟西汀能够抑制健康志愿者利培酮(为CYP3A4底物)的代谢[9-10],但其机制并不清楚。
目前认为孕烷酮X受体(pregnane X receptor, PXR)是调节肝脏药物代谢酶CYP3A4(CYP3A11,小鼠)和羧酸酯酶(CES1,CES2;或Ces1d,Ces1e,小鼠)的重要核受体[11-12],PXR的功能增加可上调其靶基因如CYP3A4、转运体MDR1的表达[13]。当PXR被激活就可向细胞核转运,与其分子伴侣RXR(retinoid X receptor, RXR)形成异源性二聚体与相应靶基因启动子结合对靶基因起转录调节作用[14]。人软骨细胞分化表达基因(differentially expressed in chondrocytes 1,DEC1;小鼠stimulated with retinoic acid 13,Stra13)属于碱性螺旋蛋白,可调节细胞分化、淋巴细胞成熟、时钟基因以及维持代谢的平衡[15]。此外,DEC1对CYP3A4具有直接抑制作用[16]。基于以上所述,本文提出氟西汀降低小鼠药物代谢酶(Ces1d,Ces1e,CYP3A11)与其降低小鼠PXR表达和增高Stra13表达相关。本研究为临床氟西汀联合用药时的合理用药提供实验依据。
1 材料与方法
1.1 实验动物 体重22g,清洁级雄性C57BL/6N小鼠,由南京医科大学实验动物中心提供。
1.2 药品 氟西汀、脂多糖(Lipopolysaccharides, LPS)和4-硝基苯基乙酸酯(para-nitrophenylacetate)购自美国 Sigma 公司; Ces1e抗体购自英国Abcam公司;Ces1d、Stra13抗体购自美国Abgent公司;β-actin抗体购自美国Bioworld公司;辣根过氧化物酶标记的二抗和BCA试剂盒购自美国Pierce公司;P450-GloTMCYP3A4 酶活性试剂盒购自美国Promega公司; ECL化学发光检测试剂盒购自美国Biouniquer公司;其余试剂均为分析纯级别。
1.3 实验动物及处理 雄性C57BL/6N 小鼠(8~10 周龄,21~22 g),实验前动物适应性饲养1 周,自由饮食,维持12 h 光照和12 h 黑暗的昼夜节律,温度:20~25 ℃,湿度:50% ± 5%。将小鼠随机分为4组,即对照组(CON)、氟西汀低剂量组(FLX-L)、氟西汀高剂量组(FLX-H)和脂多糖组(LPS)每组8 只,对照组经腹腔注射等容积生理盐水,氟西汀低剂量和氟西汀高剂量组分别注射10 mg/kg或20mg/kg的氟西汀。LPS组注射2 mg/kg LPS,本研究小组和其他研究组已经报道,LPS可降低肝脏羧酸酯酶(CESs)、细胞色素P450 3A等药物代谢酶的表达[17-18],因此,以2mg/kg LPS作为阳性对照。
1.4 组织总蛋白提取和Western Bolt分析 小鼠麻醉后用冰冷的无菌生理盐水进行肝脏灌流清除血液,取肝脏组织。称重后加入RIPA 裂解液超声破碎提取肝脏组织总蛋白。4 ℃ 12 000 r /min离心15 min 后取上清,BCA 法进行蛋白质定量。取20 μg 肝脏组织蛋白样品用7.5% SDS-PAGE 胶电泳分离。将蛋白电转至PVDF膜上,5%脱脂奶粉封闭1 h,加稀释的一抗室温孵育2 h。TBST 洗膜3 次,每次10 min。加二抗室温孵育1 h,TBST 洗膜3 次,每次10 min,加入ECL 化学发光液检测目的蛋白条带,GAPDH为内参。
1.5 酶活性测定 水解酶活性测定:在1 mL比色皿中,1 mL体积系统中,100 mM磷酸钾缓冲液(pH 7.4)含10 μg 的肝组织蛋白1 mM的4-硝基苯基乙酸酯(底物)在室温中反应,其水解速率用分光光度计(400 nm)记录。1 mL比色皿在400 nm的消光系数是13/mM/cm计算出总的水解酶活性,同时设计包括没有蛋白和没有底物等阴性对照排除系统误差。
CYP3A4活性测定:严格按照美国Promega 的CYP3A4 酶活性测定试剂盒说明书操作,最终反应产物光密度由全波长酶标仪扫描(Thermo Scientific),样品相对CYP3A4活性为样品CYP3A4活性/对照组CYP3A4活性×100%,同时设计包括没有蛋白和没有底物等阴性对照排除系统误差。

2 结果
2.1 氟西汀抑制小鼠肝脏总水解活性,抑制肝组织Ces1d、Ces1e的蛋白表达 小鼠连续给予氟西汀3 d后,肝组织总水解酶活性明显降低(P<0.05,见图1A),同时氟西汀能浓度依赖性抑制肝组织Ces1d、Ces1e的蛋白表达(P<0.05、0.01,见图1B、1C),结果提示氟西汀抑制肝组织水解酶活性是由于其抑制羧酸酯酶Ces1d、Ces1e的蛋白表达。本实验系统中, LPS抑制小鼠肝脏Ces1d和Ces1e的表达及其水解活性降低,说明实验系统状态正常。
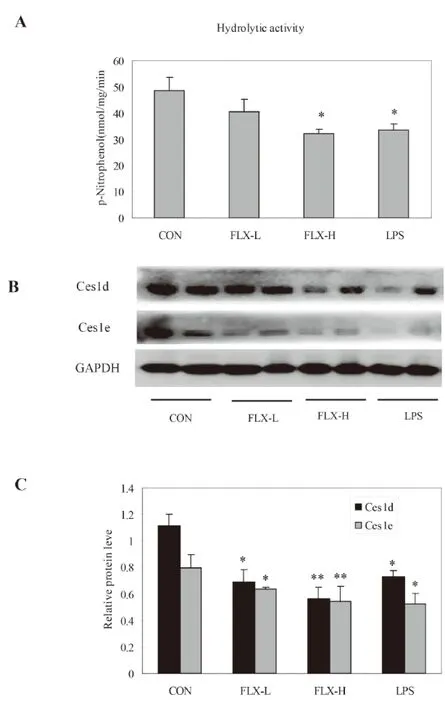
A:氟西汀对小鼠肝脏水解酶活性的影响;B:氟西汀对小鼠肝脏羧酸酯酶(Ces1d, Ces1e)蛋白表达的影响;C:Ces1d或Ces1e/GAPDH的灰度扫描密度:CON 表示对照组,FLX-L表示FLX 10 mg/kg,FLX-H表示FLX 20 mg/kg,LPS表示2 mg/kg,*P<0.05, ** P<0.01,VS对照组。
2.2 氟西汀抑制小鼠肝脏CYP3A11活性,并抑制CYP3A11的蛋白表达 小鼠连续给予氟西汀3d后,肝组织CYP3A11活性明显降低(P<0.05、0.01,见图2A),同时发现氟西汀能浓度依赖性抑制肝组织CYP3A11的蛋白表达(P<0.01,见图2B、C),结果提示氟西汀抑制肝组织氧化酶活性是由于其抑制CYP3A11的蛋白表达。本实验系统中,LPS抑制小鼠肝脏CYP3A11的表达及其氧化活性降低,说明实验系统状态正常。

A:氟西汀对小鼠肝脏CYP3A11活性的影响;B:氟西汀对小鼠肝脏CYP3A11蛋白表达的影响;C:CYP3A11/GAPDH的灰度扫描密度:CON表示对照组,FLX-L表示FLX10 mg/kg,FLX-H表示FLX20 mg/kg,LPS表示2 mg/kg,*P<0.05,**P<0.01,VS对组。
2.3 氟西汀抑制小鼠肝脏组织核受体PXR的蛋白表达 为了探讨氟西汀对肝脏药物代谢酶Ces1d、Ces1e和CYP3A11抑制作用的作用机制,我们检测调节药物代谢酶的主要核受体PXR的表达情况,结果发现:氟西汀在降低药物代谢酶的同时,能浓度依赖性抑制PXR的表达(P<0.05、0.01,见图3)。

A:氟西汀对小鼠肝脏PXR蛋白表达的影响;B:PXR/GAPDH的灰度扫描密度:CON表示对照组,FLX-L表示FLX10 mg/kg,FLX-H表示FLX20 mg/kg,LPS表示2 mg/kg,*P<0.05,**P<0.01,VS对照组。
2.4 氟西汀抑制小鼠肝脏组织转录因子 随后我们检测了小鼠肝脏Stra13(DEC1)的表达情况,结果发现:氟西汀在降低药物代谢酶的同时,能浓度依赖性增加Stra13的表达(P<0.05、0.01,见图4)。
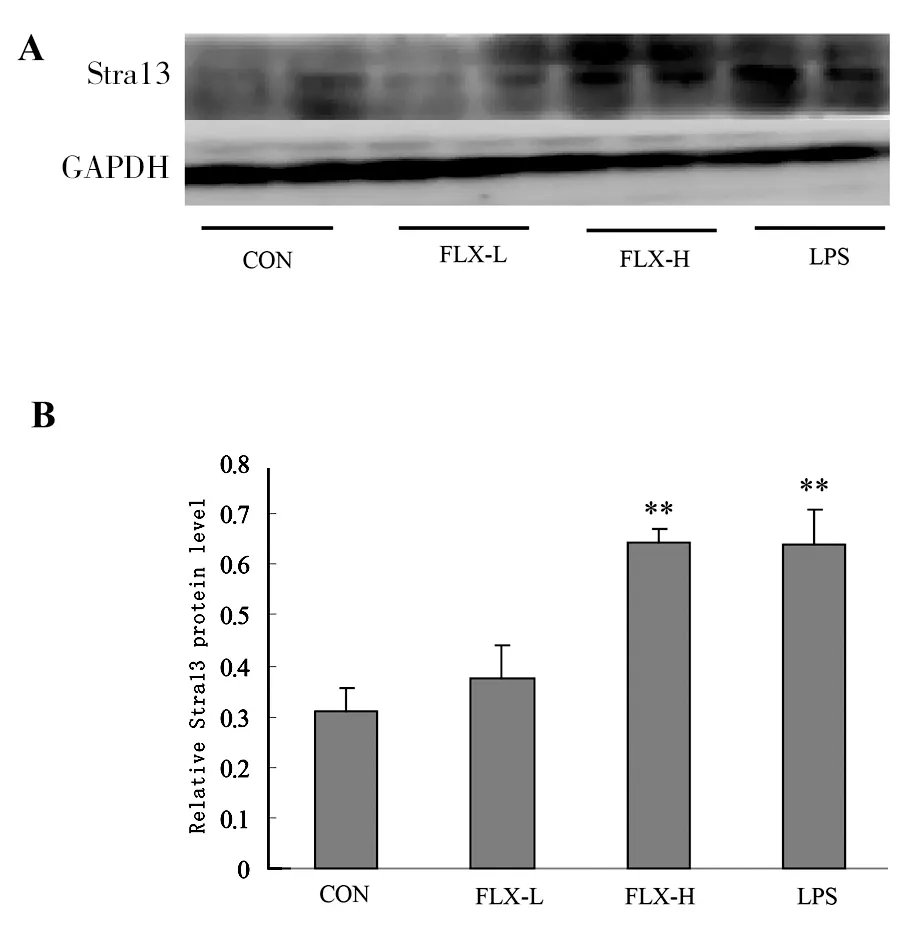
A:氟西汀对小鼠肝脏Stra13蛋白表达的影响;B:Stra13/GAPDH的灰度扫描密度:CON表示对照组,FLX-L表示FLX10 mg/kg,FLX-H表示FLX20 mg/kg,LPS表示2 mg/kg,*P<0.05,**P<0.01,VS对照组。
3 讨论
氟西汀(又名百忧解)是选择性5-羟色胺再摄取抑制类抗抑郁药中临床应用最广泛的药物,尽管近年来出现了各种新型药物,但由于高知名度及售价的低廉性,使得氟西汀仍然是广大抑郁症患者的首选用药[8]。在临床实验发现,氟西汀能减慢抗精神病药物利培酮(为CYP3A4底物)的代谢[9-10]。提示氟西汀对肝脏药物代谢酶有一定影响,但其机制并不清楚。本研究发现,氟西汀能抑制小鼠肝脏羧酸酯酶(Ces1d,Ces1e)和CYP3A11(CYP3A4)的表达及活性。尽管人体存在多种羧酸酯酶,但人肝脏主要有两种羧酸酯酶CES1(Ces1d)和CES2(Ces1e),承担多种酯类药物的水解,其功能可以激活前药而激活或增加药物作用,例如CES1(Ces1d)使抗病毒感冒的前药激活奥斯他韦激活,或降低药物毒性,如CES1(Ces1d)可水解氯比格雷使其失活,其毒性降低[19-20]。CYP3A4(CYP3A11)是肝脏CYPs最丰富和最重要的药物代谢酶,可代谢60%的临床现存处方药物[21-22]。因此,这3种肝脏主要代谢酶承担多种药物和外源性物质代谢,具有重要的生理和药理学意义。
PXR为公认的排除外来物质基因包括药物代谢酶和转运体的关键调节因子[23-24],为了阐明PXR是否在氟西汀所致的小鼠肝脏羧酸酯酶(Ces1d,Ces1e)和CYP3A11(CYP3A4)的表达及活性发挥作用,我们检测小鼠肝脏PXR的表达,发现氟西汀可浓度依赖性抑制PXR表达。提示氟西汀所致小鼠肝脏羧酸酯酶(Ces1d,Ces1e)和CYP3A11(CYP3A4)降低可能与PXR表达降低有关。
人DEC1,小鼠Stra13属于基本螺旋环螺旋结构蛋白(basic helix-loop-helix protein,bHLH),参与多种细胞功能,如细胞的增值和分化、淋巴细胞的成熟以及机体代谢,此外,Stra13(DEC1)与CYP3A4启动子近端反应元件结合而负性调节CYP3A4(CYP3A11)的表达[16],因此,我们检测了小鼠肝脏Stra13(DEC1)的表达,结果发现氟西汀能够增加小鼠肝脏Stra13蛋白水平表达,提示氟西汀抑制小鼠肝脏药物代谢酶的表达除了与其降低PXR表达以外,还与其增加Stra13(DEC1)相关。
综上所述,本研究发现氟西汀降低小鼠肝脏羧酸酯酶(Ces1d,Ces1e)和CYP3A11(CYP3A4)的表达及活性,其机制可能与氟西汀降低小鼠肝脏PXR的表达和增加Stra13(DEC1)表达有关,氟西汀如何抑制小鼠肝脏PXR表达以及如何增加Stra13表达的机制有待进一步研究。研究结果为临床合理使用氟西汀尤其是合理的与其他药物联合应用提供实验依据。
[1] Parkinson A.Biotransformation of xenobiotics, in the casarett and doull's Toxicology:the basic science of poisons (Klaassen, CD ed)[M].New York: McGraw-Hill Companies, 2001:139-162.
[2] Satoh T,Hosokawa M. Structure, function and regulation of carboxylesterases[J].Chem Biol Interact, 2006, 162(3):195-211.
[3] Ko K W, Erickson B, Lehner R. Es-x/Ces1 prevents triacylglycerol accumulation in McArdle-RH7777 hepatocytes[J].Biochim Biophys Acta, 2009, 1791(12): 1133-1143.
[4] Cooper B W, Cho T M, Tompson P M, et al. Phthalate induction of CYP3A4 is dependent on glucocorticoid regulation of PXR expression[J].Toxicol Sci, 2008, 103(2): 268-277.
[5] Shou M, Havashi M, Pan Y,et al. Modeling, prediction, and in vitro in vivo correlation of CYP3A4 induction[J].Drug Metab Dispos, 2008,36(11):2355-2370.
[6] Lopez A D, Mathers C D, Ezzati M,et al. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data[J].Lancet, 2006, 367(9524): 1747-1757.
[7] Freeborough A, Kimpton J. Discovering new genetic and psychosocial pathways in Major Depressive Disorder: the NewMood project[J].Psychiatr Danub, 2011, 23(Suppl 1):138-141.
[8] Wong D T, Perry K W, Bymaster F P. Case history: the discovery of fluoxetine hydrochloride (Prozac)[J].Nat Rev Drug Discov, 2005, 4(9): 764-774.
[9] Spina E, Avenoso A, Scordo M G,et al. Inhibition of risperidone metabolism by fluoxetine in patients with schizophrenia: a clinically relevant pharmacokinetic drug interaction[J].J Clin Psychopharmacol, 2002, 22(4): 419-423.
[10] DeVane C L, Donovan J L, Liston H L,et al. Comparative CYP3A4 inhibitory effects of venlafaxine, fluoxetine, sertraline, and nefazodone in healthy volunteers[J].J Clin Psychopharmacol, 2004,24(1): 4-10.
[11] Staudinger J L, Xu C, Cui Y J,et al.Nuclear receptor-mediated regulation of carboxylesterase expression and activity[J].Expert Opin Drug Metab Toxicol,2010,6(3):261-271.
[12] Deng R, Xu C, Chen X,et al. Resveratrol suppresses the inducible expression of CYP3A4 through the pregnane X receptor[J].J Pharmacol Sci,2014,126(2):146-154.
[13] Liu F J, Song X, Yang D,et al.The far and distal enhancers in the CYP3A4 gene co-ordinate the proximal promoter in responding similarly to the pregnane X receptor but differentially to hepatocyte nuclear factor-4alpha[J].Biochem J, 2008, 409(1): 243-250.
[14] Gu X, Ke S, Liu D,et al. Role of NF-kappaB in regulation of PXR-mediated gene expression: a mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents[J].J Biol Chem, 2006, 281(26):17882-17889.
[15] Iizuka K, Horikawa Y. Regulation of lipogenesis via BHLHB2/DEC1 and ChREBP feedback looping[J].Biochem Biophys Res Commun, 2008, 374(1): 95-100.
[16] Mao Z, Luan X F, Cao G,et al. DEC1 binding to the proximal promoter of CYP3A4 ascribes to the downregulation of CYP3A4 expression by IL-6 in primary human hepatocytes[J].Biochem Pharmacol, 2012, 84(5): 701-711.
[17] Mao Z, Li Y, Peng Y,et al. Lipopolysaccharide down-regulates carbolesterases 1 and 2 and reduces hydrolysis activity in vitro and in vivo via p38MAPK-NF-κB pathway[J].Toxicol Lett, 2011, 201(3):213-220.
[18] Zhang Y, Li Y, Li Q. Inhibition of cytochrome P450 3A in rat liver by the Diorganotin (IV) compound di-n-Butyl-di-(4-chlorobenzo-hydroxamato)tin (IV) and Its Probable Mechanism[J].Molecules,2012,17(9):10994-1009.
[19] Tang M, Mukundan M, Yang J,et al. Antiplatelet agents aspirin and clopidogrel are hydrolyzed by distinct carboxylesterases and clopidogrel is transesterified in the presence of ethyl alcohol[J].J Pharmacol Exp Ther, 2006, 319(3):1467-1476.
[20] Hu H J, Wang X, Gawronski B E,et al. Carboxylesterase 1 as a determinant of clopidogrel metabolism and activation[J].J Pharmacol Exp Ther, 2013,344(3):665-672.
[21] Lonsdale R, Rouse S L, Sansom M S, et al.A multiscale approach to modelling drug metabolism by membrane-bound cytochrome P450 enzymes[J].PLoS Comput Biol,2014,10(7):e1003714.
[22] Mitsui T, Nemoto T, Miyake T,et al. A useful model capable of predicting the clearance of cytochrome 3A4 (CYP3A4) substrates in humans: validity of CYP3A4 transgenic mice lacking their own Cyp3a enzymes[J].Drug Metab Dispos, 2014,42(9):1540-1547.
[23] Kojima K, Nagata K, Matsubara T,et al. Broad but distinct role of pregnane x receptor on the expression of individual cytochrome p450s in human hepatocytes[J].Drug Metab Pharmacokinet, 2007, 22(4): 276-286.
[24] Healan-Greenberg C, Waring J F, Kempf D J,et al. A human immunodeficiency virus protease inhibitor is a novel functional inhibitor of human pregnane X receptor[J].Drug Metab Dispos, 2008, 36(3):500-507.
[收稿2014-12-10;修回2015-01-04]
(编辑:谭秀荣)
The effect of fluoxetin on Ces1d,Ces1e and CYP3A11 in the mouse liver
ShangWei,WangYuwen,ChuDongbao,XiongJing,LiuWei,LiuJie,ChenRuini,YangJian
(Department of Pharmacology, Nanjing Medical University,Nanjing Jiangsu 210029,China)
Objective To study the effect of fluoxetin on drug metabolism enzymes in the mouse liver.Methods C57BL/6N mice were divided into four groups. The mice were injected intraperitoneally with equal volume of saline, 10,20 mg/kg fluoxetin, or 2 mg/kg LPS, respectively for three days. The livers were perfused to remove blood and Collected for enzyme assay and Western blot analysis.Results Fluoxetin represses the expression of Ces1e, Ces1 d and hydrolytic activity, and represses CYP3A11 and its activity in the mouse liver. Meanwhile, fluoxetin decreases PXR expression, which is an important regulator of drug metabolism enzymes, whereas increases Stra13 (DEC1) expression, which down-regulates CYP3A11 (CYP3A4).Conclusion The repression of the drug metabolism enzymes(Ces1 d、Ces1e、CYP3A11) induced by fluoxetin in the mouse liver is related to the decrease of PXR and increase of Stra13 (DEC1).
Fluoxetin; drug metabolism enzymes; PXR; Stra13
国家自然科学基金资助项目(NO:81173128,NO:81373443,NO:81302855,NO:81102457);江苏省教育厅重大项目(NO:13KJA310003);高等学校博士学科点专项科研基金资助项目(NO:20123234110005)。
杨俭,女,博士,教授,博士生导师,研究方向:临床药理学,E-mail:jianyang@njmu.edu.cn。
R749.8
A
1000-2715(2015)01-0015-05
猜你喜欢
轻工学报(2022年3期)2022-06-22
中国现代医生(2022年6期)2022-04-23
食品科学(2022年6期)2022-03-30
建材发展导向(2021年7期)2021-07-16
建材发展导向(2021年24期)2021-02-12
医药前沿(2020年26期)2020-12-02
建材发展导向(2019年5期)2019-09-09
世界农药(2019年2期)2019-07-13
分析化学(2017年12期)2017-12-25
心脑血管病防治(2016年6期)2017-01-16
