
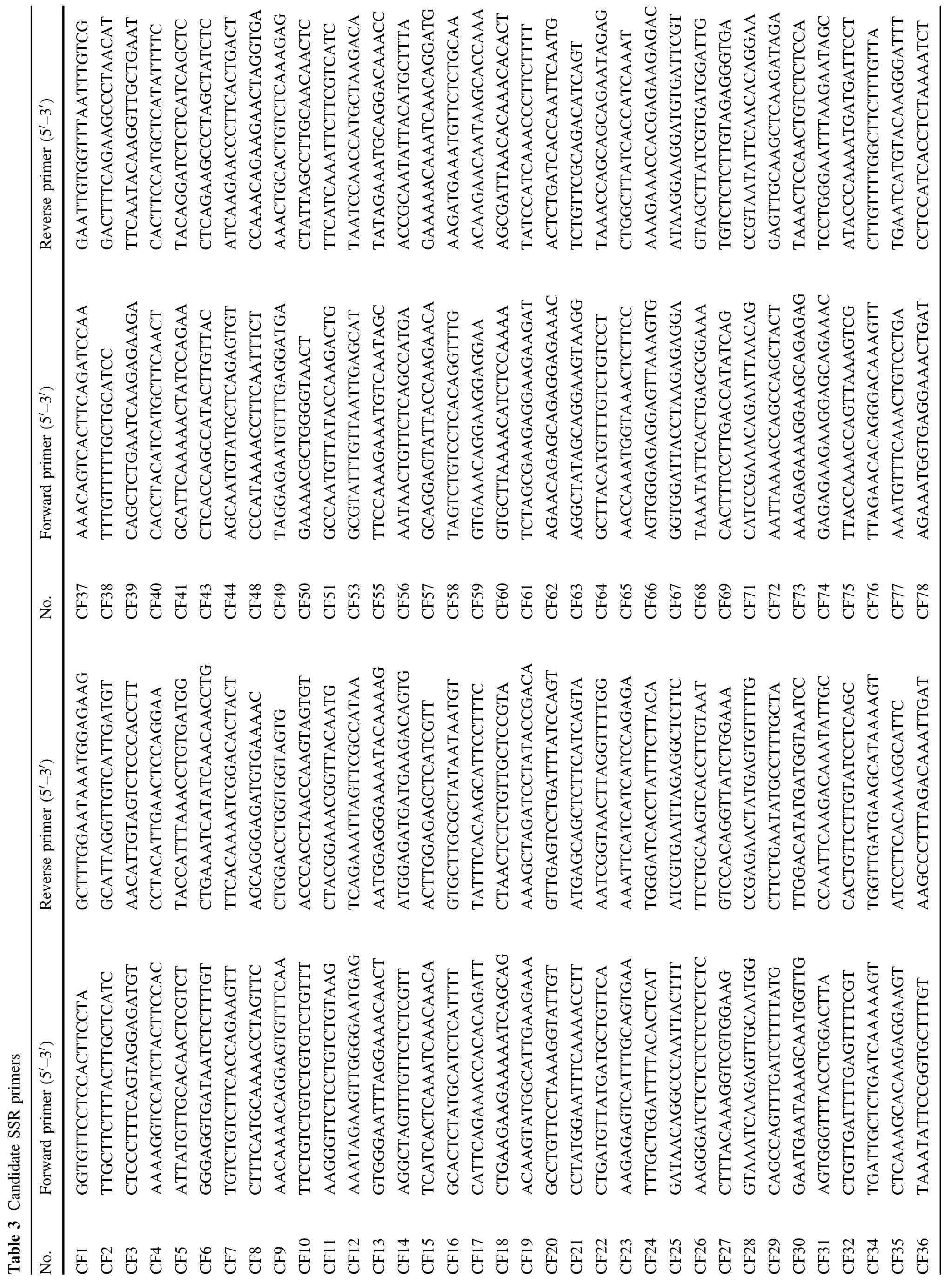
Exploit SSR in Cryptromeria fortunei and application in genetic diversity analysis
2015-06-09 18:06••
Journal of Forestry Research 2015年4期
••
ORIGINAL PAPER
Exploit SSR in Cryptromeria fortunei and application in genetic diversity analysis
Yongquan Lu1•Jingli Zhang1•Zaikang Tong1
©Northeast Forestry University and Springer-Verlag Berlin Heidelberg 2015
Cryptromeria fortuneiis one of the main forest plantation species in the subtropical high altitude areas in China.In this paper we collected 49C.fortuneigermplasm resources and provides a study of the utility of freely availableC.japonicaEST resources for the development of markers necessary for genetic diversity analyses ofC. fortunei.By screening 24,299 EST sequences fromC. japonicawith SSR Finder,we identif i ed 2384 ESTs carrying 2783 SSR motifs.We successfully obtained 364 (15%)primers from 2419 putative SSR loci.Of the 80 candidate SSR markers tested,70(87.5%)yielded stable and clear PCR products.With those primers,the genetic diversity of 49C.fortuneiwe collected was studied.The results showed that 18 primers yield polymorphism within these accessions.These 18 primers generated 48 scorable SSR loci and the average number of polymorphic SSR loci per primer was 2.7.The PIC value varied from 0.375 to 0.8101,with the average of 0.4780.The Shannon index is 0.5718,and the value of the observed number of alleles and effective number of alleles are 1.9167 and 1.7289, respectively.The genetic coeff i cient of these 49 accessions varied from 0.28 to 0.87.According to the genetic distances,a cluster tree was constructed.At genetic coeff i cient of 0.60,these 49 accessions can group into 3:group I contains only FJ-laizhou accession,and group II contains 2 accessions from FJ-layang,and the other one group contains mixed accessions.At genetic coeff i cient of 0.68,the former group II was constructed into 7 subgroups,with the fi rst 3 subgroups contain 16 accessions in which 11(69%) are from Fujian province,and the later 4 subgroup contain 31 accessions in which 20(65%)were from Zhejiang province.
Cryptromeria fortunei⋅EST–SSR⋅Germplasm collection⋅Genetic diversity
Introduction
TheCryptromeria fortuneiwithin the Taxodiaceae family are fast-growing evergreen trees.It is one of the main forest plantation species in the subtropical high-altitude areas in China.Taxodiaceae families are relict plants from Cretaceous period.The family was an important component in forest vegetation of the northern hemisphere from the late Cretaceous to the mid-tertiary eras approximately 115–30 million years ago.In the late tertiary and pleistocene,however,the family underwent a widespread reduction resulting in the present-day relictual genera with restricted distributions(Sehlarbaum and Tsuehiya 1984).
At precent,theCryptromeriais olictypic genera that consists of only 2 species,includingC.fortunei and Cryptromeria japonica.The natural distribution ofC.fortuneiis extremely narrow,consisting of interrupted or isolated distribution in the Wuyishan and Tianmu Mountains in China(Wang et al.2007).In recent years,driven by economic interests and inf l uenced by environmental factors such as insect damage,C.fortuneiresources have sharply fallen.Plant genetic resources are an importantmaterial basis for breeding and species diversity protection. Therefore,it is urgent to collect and protectC.fortuneiresources.Genetic diversity generally refers to the genetic diversity within a species.The level of genetic diversity directly affects the sustainable development of a population.Molecular markers are an important tool to study genetic diversity.The polymorphisms revealed by molecular markers have no impact by external environment and internal development stage,and therefore are suitable for genetic diversity evaluation.Expressed sequence tag (ESTs)are generated from single-pass sequencing of randomly picked cDNA clones(Adams et al.1991).The EST approach and subsequent gene-expression prof i ling(cDNA microarrays)have proven to eff i ciently evaluate genetic diversity(Enorki et al.2002,Riar et al.2011).ESTs are necessary for developing EST–SSR markers.
With the recent progress made in the large-scale function genome sequencing project,thousands of open data sets have been generated.So far,however,there remains no EST data,nor EST–SSR markers,inC.fortunei.According to Yang et al.(2007),EST-derived markers are likely to be conserved across a broader taxonomy than any other sort of marker.For those species with scarce sequence information,it is may be feasible to develop EST–SSR markers using EST data for closely related taxa(Lu et al.2013).C. japonica,which has lots of sequencing and biological information in public databases,is closely related toC. fortunei(Futamura et al.2008;Tani et al.2003,2004).
Moriguchi et al.(2009)exploited EST–SSR markers inC.japonicaand 27 EST–SSR marker sequences have been published.In theory,those markers might be used in genetic diversity analysis inC.fortunei;however,those markers exploited inC.japonicaare not enough to meet the need ofC.fortuneistudies.To meet this need,we provide a study of the utility of freely availableC.japonicaEST resources for the development of markers necessary for genetic diversity analyses ofC.fortunei.
Materials and methods
Collection of germplasm resources
In the spring of 2013,49 samples ofC.fortuneigermplasm were collected from Zhejiang and Fujian provinces,China, respectively(Table 1).The new tender branches ofC. fortuneiwere cut and brought back to the Laoshan forest farm,located in Zhejiang province.The tender branches were grafted cambium to cambium.After 2 months,the shoots made a strong union with the stock and the new leaves emerged.
The new leaves fromC.fortuneiwere processed for DNA isolation.Total genomic DNA was isolated from 200 mg of fresh leaf using the CTAB method(Murray and Thompson 1980).DNA quality was detected using a UV spectrophotometer and DNA in accordance with the standard was used in following experiments.
Search of putative SSR
In total,24,299 EST sequences ofC.japonica,released by the plant GDB(http://www.plantgdb.org)were examined. We used simple sequence repeat identif i cation tool (SSRIT)(http://www.gramene.org/db/searches/ssrtool)to search SSR among these ESTs.Those ESTs including a 2-4 bp repeat motifs were selected as putative SSR.To develop candidate SSR markers from the SSRs identif i ed with SSR Finder,we designed PCR primers based on fl anking regions on the EST sequences using ePrimer3 (Rozen and Skaletsky 2000).To increase the quality and usability of the silicon exploited SSR markers,we required exact matches between primers and templates,and the product sizes were between 100 and 300 bp.
Verif i cation of SSR markers inC.fortunei
Eighty primers were selected and synthesized by Songgong Engineering&Technology Company in China.ThreeC. fortuneisamples,which represent different ecotypes,were used to verify the candidate SSR markers.PCR was performed in 20 μL reactions containing 50 ng of template DNA,0.5 μmol/L of each primer,200 μmol/L of each dNTP,1.5 mmol/L of MgCl2,1 unit of Taq polymerase, and 2 μL of 10×PCR reaction buffer.
The following PCR program was used:5 min at 95°C; 30 cycles of 30 s at 95°C;30 s at 56°C;1 min at 72°C; and 7 min at 72°C for a f i nal extension.For those primer pairs that did not generate good amplif i cation results,the initial annealing temperatures were adjusted from 55 to 60°C.Each of the primer pairs was tested at least twice to conf i rm the repeatability of the observed bands in each sample.PCR products were separated on 0.8%agarose gel.Gels were stained with ethidium bromide for visualizing DNA bands.
Diversity analysis ofC.fortunei
Primer pairs that generate good amplif i cation results were used for analysis of the diversity of 49C.fortunei.PCR reactions were the same as mentioned above.Only clear and reproducible PCR products were separated on 6% non-denaturing polyacrylamide gel(80 Volts,2.5 h).Gels were silver stained following the procedure in Xu et al. (2002).Bands were scored as present(1)or absent(0) directly from the gel.Based on the PCR data,we evaluated the allelic diversity of each SSR marker using thepolymorphism information content(PIC)value,def i ned asis the frequency of the ith marker.POPGENE 32 was used to calculate the observed number of alleles(Na),effective number of alleles(Ne), and Shannon’s information index(I).Genetic relationships and their grouping on the basis of the genetic distance yielded SSR markers that were assessed using Nei’s coeff i cients(Nei and Li 1979).A dendrogram was generated from the genetic distance matrix using the unweighted pair-group method with arithmetic averages UPGMA, using the NEIGHBOR model of the NTSYS Version 2.1 software(Ueno et al.2012).

Table 1 Collection sites of Cryptromeria fortunei germplasm resources
Results and discussion
Candidate SSR markers
By screening 24,299 EST sequences fromC.japonicawith SSR Finder,we identif i ed 2384 ESTs carrying 2783 SSR motifs.These results showed that 9.8%ESTs have SSR. Among these ESTs,2068 contain one SSRmotif,252 contain 2 SSR motifs,46 contain 3 SSR motifs,17 contain 4 SSR motifs,and 1 contains 5 SSR motifs.Tri-SSRs were the most common SSRs,counting for 48.47%,and the most common motifwas GAA(Fig.1).The frequency ofthe otherSSRtypes and their most common motifs are in Table 2.
Exploit SSR markers
We successfully obtained 364(15%)primers from 2419 putative SSR loci.Those that did not yield a product were the result of too short sequences f l anking the SSRs.The candidate primers were selected and named with the abbreviation CF(for primerC.fortunei)followed by a unique number(e.g.,Cf 10).We selected 80 candidate SSR markers for further testing.
Experimental tests of candidate SSR markers
All primers were tested on three differentC.fortuneiDNA. Of the 80 candidate SSR markers tested,70(87.5%) yielded stable and clear PCR products(Fig.2).Because the primers were designed fromC.japonicaEST data,the result showed the transferability of these two species is relatively high.The primers of these newC.fortuneiSSR markers and their name are shown in Table 3.
Genetic diversity analysis ofC.fortunei
The genetic diversity of 49C.fortuneiwas studied.The results showed that 18 primers yielded polymorphism within these accessions.The information of 18 polymorphic primers can be seen in Table 4.
These 18 primers generated 48 scorable SSR loci and the average number of polymorphic SSR loci per primer was 2.7.Patterns of variation observed with the primer CF18 can be seen in Fig.3.Clearly,SSR variation existed among the 49C.fortunei.The PIC value varied from 0.375 to 0.8101,with an average of 0.4780.The Shannon index is 0.5718,and the value of Na and Ne are 1.9167 and 1.7289, respectively.
The genetic coeff i cient of the 49 accessions varied from 0.28 to 0.87.A cluster tree was constructed,according to the genetic distances(Fig.4).At genetic coeff i cient of 0.60,the 49 accessions fall into three groups:group 1 contains only FJ-laizhou accession,group II contains 2 accessions from FJ-layang,and group 3 contains mixed accessions.It was as expected that 2 accessions from FL-layang formed a group f i rst.Because there is only one accession from FJ-laizhou,determining the genetic distance of this accession is larger than the others.At genetic coeff i cient of 0.68,the former group II was constructed into 7 subgroups,with the f i rst 3 containing 16 accessions in which 11(69%)are from FJ province,and the later 4subgroups containing 31 accessions in which 20(65%) were from ZJ province.

Fig.1 Proportions of Tri-SSRs motifs within each type

Table 2 SSR number,abundance and distribution frequency of each motif

Fig.2 Candidate primers amplif i ed in C.fortunei
Discussion
EST–SSR markers inC.fortunei
EST–SSR genetic markers reside in gene sequences that can directly ref l ect aspects of variation within those genes. Therefore,the genetic relationship tree constructed with EST–SSR markers could be especially valuable for genetic studies.However,levels of polymorphism could be low because these EST–SSRs are in expressed regions that have more evolutionary conservation compared with primers derived from genome SSR(gSSR)(Xu et al.2014). However,results from poplar(Zhang et al.2011)showed that,compared with gSSR,EST–SSR markers were superior in versatility for different species of poplar,and EST–SSR loci revealed higher overall levels of genetic diversity than gSSR loci.At present,however,no EST data remains inC.fortunei.According to Yang et al.(2007),EST-derived markers are likely to be conserved across a broader taxonomic range than any other type of marker.For those species with a scarcity of sequence information,EST data from closely related species could be used.Therefore,we usedC.japonicaEST data to exploit SSR markers forC. fortunei.On the other hand,our results showed that thosesequence data were transferable among these 2 species. Ueno et al.(2012)developed an open framework for the analysis of microsatellites in expressed sequence tags. They used this pipeline to develop EST–SSR markers forC.japonica.However,the SSR frequency detected by their framework was lower(4.54%)than our method(9.8%). Consequently,our study developed an economic effectiveness analyzing tool for development of SSR markers inC.fortunei.
?

Table 4 EST–SSR information of 18 polymorphic primers
C.japonica and C.fortunei
There has long been argument about classif i cation of theCryptomeriagenus.Most Chinese taxonomists considerCryptomeriagenus to include two species:C.japonicaandC.fortunei(namely ChineseCryptomeria).Some others consider that ChineseC.fortuneiis a variety ofC.japonica.According to our studies,most primers(70 primers out of 80)yielded fromC.japonicaEST can be used inC. fortunei,which indicated that the nucleic acid sequence of the two species has higher similarity.But on the other hand,10 primers out of 80 EST–SSR fromC.japonicayielded no ampli fi cation products inC.fortunei,suggesting that the two species in the DNA sequence also developed a certain degree of differentiation.The long-term geographical separation may lead to population genetic drift, resulting in the formation of two different populations. Although the difference is relatively small,the DNA sequence differences can be inherited and generate new genotype.
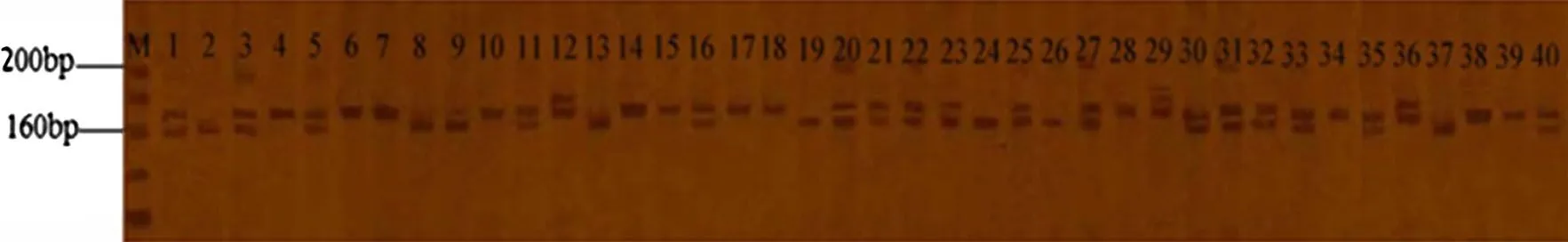
Fig.3 Polymorphisms of C.fortunei amplif i ed by CF18.Note the number correspondence to the resources number in Table 1

Fig.4 Dendrogram of 49 accessions of C.fortunei.Note the number correspondence to the resources number in Table 1
Genetic diversity ofC.fortunei
C.japonicais one of the oldest species in China.Using 18 pairs of EST–SSR primers,we detected 49 accessions ofC. fortunei.The Shannon index ofC.fortuneipopulation that we used in this study is 0.5718,which is relatively high, however large errors may exist.In this study,C.fortuneiresources were collected from Zhejiang and Fujian.Some provenances,such as Fujian-Laizhou and Fujian-Layang, have few liveC.fortuneitrees,so we only able to obtain one or two samples.This may be the main reason for the high Shannon index.We should give more protection to theC.fortuneiresources.
In genetic diversity analysis of the 49 resources,a small amount of different geographic origins cluster together, which implies that the genetic diversity ofC.fortuneiin China is fairly low,probably because the species is windpollinated and long-lived.Takahashi et al.(2005)investigated the allelic variation and genetic structure ofC. japonica,and the results showed that no clear relationship between Nei’s genetic distances and geographical locations of the populations were found using the principal coordinate and unweighted pair-group methods with arithmetic averaging analyses.In this study,no clearly relationship was found between genetic distances and geographical locations in theC.fortuneigroup as well.Therefore,during the process of resources conservation and breeding forC. fortunei,the geographical distance,as well as genetic distance should be considered.
Adams MD,Kelley JM,Gocayne JD,Dubnick M,Polymeropoulos MH,Xiao H,Merril CR,Wu A,Olde B,Moreno RF,Kerlavage AR,McCombie WR,Venter JC(1991)Complementary DNA sequencing:expressed sequence tag and human genome project. Science 252:1651–1656
Enorki H,Sato H,Koinuma K(2002)SSR analysis of genetic diversity among maize inbred lines adapted to cold regions of Japan.Theor Appl Genet 104:1270–1277
Felsenstein J(1989)PHILIP-Phylogeny inference package.Cladistics 5:164–166
Futamura N,Totoki Y,Toyoda A,Igasaki T,Nanjo T,Seki M,Sakaki Y,Mari A,Shinozaki K,Shinohara K(2008)Characterization of expressed sequence tags from a full-length enriched cDNA library ofCryptomeria japonicamale strobili.BMC Genomics 9:383–396
Lu Y,Jia Q,Tong Z(2013)Development of amplif i ed consensus genetic markers in Taxodiaceae based onCryptomeria japonicaESTs data.J For Res 24:503–508
Moriguchi Y,Iwata H,Ujino-Ihara T(2003)Development and characterization of microsatellite markers forCryptomeria japonicaD.Don.Theor Appl Genet 106:751–758
Moriguchi Y,Ueno S,Ujino-Ihara T,Futamura N,Matsumoto A, Shinohara K,Tsumura Y(2009)Characterization of EST–SSRs fromCryptomeria japonica.Conserv Genet Resour 1:373–376
Murray MG,Thompson WF(1980)Rapid isolation of high molecular-weight plant DNA.Nucl Acids Res 8:4321–4325
Nei M,Li W(1979)Mathematical model for study the genetic variation in terms of restriction endonucleases.Proc Natl Acad Sci 74:5267–5273
Riar DS,Rustgi S,Burke IC,Gill KS,Yenish JP(2011)EST–SSR development from 5Lactucaspecies and their use in studying genetic diversity amongL.serriola biotypes.J Hered 102:17–28
Rozen S,Skaletsky H(2000)Primer3 on the WWW for general users and for biologist programmers.Methods Mol Biol 132:365–386
Sehlarbaum SE,Tsuehiya T(1984)Cytotaxonomy and phylogeny in certain species of Taxodiaceae.Syst 147:29–54
Takahashi T,Tani N,Taira H,Tsumura Y(2005)Microsatellite markers reveal high allelic variation in natural populations ofCryptomeria japonicanear refugial areas of the last glacial period.J Plant Res 118:83–90
Tani N,Takahashi T,Iwata H,Mukai Y,Ujino-Ihara T,Matsumoto A,Yoshimura K,Yoshimaru H,Tsumura Y(2003)A consensus linkage map for Sugi(Cryptomeriajaponica)from two pedigrees,based on microsatellites and expressed sequence tags. Genetics 165:1551–1568
Tani N,Takahashi T,Ujino-Ihara Y,Iwata H,Yoshimura K,Tsumura Y(2004)Development and characteristics of microsatellite markers for sugi(Cryptomeria japonicaD.Don)derived from microsatellite-enriched libraries.Ann For Sci 61:569–575
Ueno S,Moriguchi Y,Uchiyama K,Tsumura Y(2012)A second generation framework for the analysis of microsatellites in expressed sequence tags and the development of EST–SSR markers for a conifer,Cryptomeria japonica.BMC Genomics 13:136–151
Wang J,Liu J,Huang Y,Yang H(2007)The origin and natural distribution ofCryptomeria.J Sichuan For Sci Technol 28:92–94(in Chinese with an English abstract)
Xu S,Tao Y,Yang Z,Chu J(2002)A simple and rapid method used for silver staining and gel preservation.Hereditas 24:335–336(in Chinese with an English abstract)
Xu Y,Chen J,Li Y,Hong Z,Wang Y,Zhao Y,Wang X,Shi J(2014) Development of EST–SSR and genomic-SSR in Chinese f i r. J Nanjing For Univ 38(1):9–14(in Chinese with an English abstract)
Yang L,Jin G,Zhao X,Zheng Y,Xu Z,Wu W(2007)PIP:a database of potential intron polymorphism markers.Bioinformatics 23:2174–2177
Zhang Y,Peng C,Li Z,Yang Y,Hu X(2011)Genetic diversity of genomic-SSR and EST–SSR markers in interspecies of Poplar. J Northeast For Univ 39(12):8–11(in Chinese with an English abstract)
8 January 2014/Accepted:28 May 2014/Published online:25 July 2015
The online version is available at http://www.springerlink.com
Corresponding editor:Hu Yanbo
✉Yongquan Lu
luyongquan@126.com
1A Nurturing Station for the State Key Laboratory of Subtropitical Silviculture Zhejiang Agriculture and Forestry University,Lin’an 311300,China
 Journal of Forestry Research2015年4期
Journal of Forestry Research2015年4期
- Journal of Forestry Research的其它文章
- Drone remote sensing for forestry research and practices
- Life cycle environmental impact assessment of biochar-based bioenergy production and utilization in Northwestern Ontario, Canada
- Growth rates of Eucalyptus and other Australian native tree species derived from seven decades of growth monitoring
- Effect of f i rst thinning and pruning on the individual growth of Pinus patula tree species
- The inf l uence of selective cutting of mixed Korean pine(Pinus koraiensis Sieb.et Zucc.)and broad-leaf forest on rare species distribution patterns and spatial correlation in Northeast China
- Modeling forest f i res in Mazandaran Province,Iran