
添加AOT的微乳液法制备纳米SnO2及其表征
2015-06-01 10:17:10刘雪华
福州大学学报(自然科学版) 2015年4期
刘雪华, 唐 电
(1. 福建工程学院材料科学与工程学院, 福建 福州 350118; 2. 福建省新材料制备与成形技术重点实验室, 福建 福州 350116; 3. 福州大学材料科学与工程学院, 福建 福州 350116)
添加AOT的微乳液法制备纳米SnO2及其表征
刘雪华1, 2, 3, 唐 电2, 3
(1. 福建工程学院材料科学与工程学院, 福建 福州 350118; 2. 福建省新材料制备与成形技术重点实验室, 福建 福州 350116; 3. 福州大学材料科学与工程学院, 福建 福州 350116)
采用AOT( 二-乙基己基琥珀酸酯磺酸钠)为表面活性剂, 以正庚烷为油相, 以四氯化锡为前驱物, 采用油包水型微乳液法制备了纳米SnO2颗粒, 并借助XRD、 DTA-TG、 FTIR、 SEM等手段系统研究了制备温度、 AOT质量、 溶液pH值等因素对产物性能的影响. 本实验体系中的最佳工艺参数为: pH=8, 水-表面活性剂摩尔比1 ∶2, AOT质量0.6 g, 锻烧温度600 ℃, 保温时间2 h. 在最佳试验条件下所制备的SnO2粉末平均粒径为4.1 nm, 较常规沉淀法制备的SnO2颗粒细小且团聚少, 具有更好的性能.
AOT; 微乳液法; SnO2; 纳米; 制备; 表征
0 引言
氧化锡(SnO2)是一种十分重要的n型宽带隙(3.6 eV)半导体无机材料, 在染料敏化太阳能电池、 新型气敏材料、 光致发光材料、 光催化材料、 红外反射玻璃、 抗静电涂层材料、 透明电极材料等领域都有广泛应用[1-7]. 氧化锡半导体纳米晶的性质和应用取决于其晶型、 形貌、 尺寸和结晶度等关键因素[8]. 实现微观结构调控可以更深入地研究氧化锡半导体纳米晶的微观结构和性能之间的构效关系, 更好地拓展其潜在的应用, 因此, 研究氧化锡纳米晶的可调控制备技术具有重要的意义[8].
在各种各样的制备方法中, 微乳液法以其独特优点引起人们的注意. 微乳液法是将金属盐和一定的沉淀剂形成微乳状液, 在较小的区域(称为“微反应器”)内控制胶粒成核和生长, 最后经热处理得到超细粒子的一种方法. 该方法最大的优点在于能够很好地控制颗粒的形核和生长过程, 从而控制其晶粒尺寸、形态、 均匀性及其比表面积[9]等, 避免在煅烧工艺过程中基于奥斯瓦尔德原理而出现的严重团聚现象, 为得到均匀分散、 晶粒细小的SnO2纳米材料制备, 开辟了一种新的思路.
国内外学者采用微乳液法制备无机金属氧化物材料的研究不过短短十数年, 取得了一些成果[10-12], 但对其形成机理、 过程、 工艺的研究尚未透彻, 而将之应用于SnO2材料的研究更是不够充分, 采用的微乳液体系各不相同, 制备产物的平均尺寸分布从3 nm[11]到 35.34~58.86 nm[12], 波动较大. 其中韩国的Song等[10]、 印度的Meitram等[13]采用AOT (2-乙基己基磺基琥珀酸钠)和正庚烷体系来制备纳米SnO2粉末, 获得小于40 nm的细小产物. 但该体系中的一些关键影响因素, 如: 反应物浓度、 制备温度、 溶液pH值等工艺条件的影响规律尚未进行系统研究, 在这种背景下, 本研究选择微乳液法来制备SnO2纳米晶粉末, 结合XRD、 FTIR、 TGA、 SEM等检测方法与手段, 探讨其制备时AOT-正庚烷体系中工艺因素的影响规律, 实现可调控制备, 为工业生产SnO2纳米材料打下良好的理论基础. 另外, 将此方法获得的氧化锡纳米粉末与传统的沉淀法相比较, 研究在相同的热处理温度下, 两种方法制备的SnO2材料所具有的优缺点.
1 实验部分
1.1 制备方法
采用两种方法制备二氧化锡纳米颗粒: ① 传统的化学沉淀法; ② 微乳液法. 在化学沉淀法中, 将28%(质量分数)的氨水溶液在均匀搅拌中缓慢加入0.1 mol·L-1的SnCl4·5H2O原料溶液至一定pH值, 使之产生均匀沉淀物. 之后将沉淀物进行抽滤, 90 ℃烘干1 h, 并在600 ℃下煅烧0.5 h.

表1 微乳液配方
在微乳液法中, 以AOT为表面活性剂, 正庚烷为油相, 将锡盐和沉淀剂氨水溶液分别配置成两种确定成分的微乳液体系: 微乳液I和微乳液II, 然后在匀速搅拌中将其混合进行3 h的沉淀反应, 以保证溶液浓度的相对恒定. 具体配方如表1. 反应结束后得到悬浊液, 将之在10 000 r·min-1转速下离心10 min, 得到沉淀物于90 ℃干燥1 h后, 在不同温度下煅烧2 h.
1.2 表征
采用STA-449C型热重-差热分析仪在空气气氛下对干燥后产物进行TG-DTA分析, 升温速率20 ℃·min-1, 从室温扫描到900 ℃. XRD分析采用德国布鲁克公司Advance D8型X射线衍射仪, Cu靶,λ=0.154 056 nm, 扫描速率为4°·min-1, 工作管压40 kV, 工作电流40 mA. 采用S-3400N型扫描电镜对产物进行显微形貌分析. 红外分析采用Nicolet FT-IR 6700型傅里叶变换红外光谱仪, 扫描范围: 400~4 000 cm-1, KBr压片, 室温条件下记录. 产率计算采用称重法.
2 结果与分析
2.1 热分析

图1 SnO2先驱体粉末的TGA-DSC曲线Fig.1 TGA-DSC curves of SnO2 precursor powders
根据前期试验, 保持AOT质量为0.6 g, 保温时间2 h, pH=8~9, 水-表面活性剂摩尔比为1 ∶2, 经混合搅拌, 反应3 h后离心分离, 90 ℃干燥2 h后可得到产物粉末. 此粉末的TGA-DSC曲线如图1所示. 由图1可知, 由室温升至220 ℃有明显的失重台阶, 约有9.30%的失重, 应为样品物理吸附水的挥发, 该阶段的失重程度取决于干燥后残留在样品中的吸附水含量. 继续升高温度, TGA曲线在220~550 ℃时, 产生一个极快失重台阶, 失重约有40.21%, 对应于残留NH3·H2O、 有机物的分解与挥发, 以及Sn(OH)4脱去化学结合水(结构水)分解为SnO2的过程. 在600 ℃以后, 样品质量略有变化, 同时DSC曲线上在同样温度范围出现放热峰, 此变化是纳米微晶中无规则分布原子的有序化造成的. 此后随着热处理温度的升高, DSC曲线上没有其它明显的峰出现, TGA曲线波动同样趋于平缓, 表明粉末已完成由非晶态向晶态的结构转变, 涉及重量变化的反应基本完成. 故可以初步确定, 600 ℃煅烧即可得到结晶较好的SnO2颗粒.
2.2 XRD分析2.2.1 AOT含量的影响

表2 不同AOT含量合成SnO2的平均
根据前期研究结果, 保持热处理温度为600 ℃、 保温时间为2 h, pH=8~9, 水-表面活性剂摩尔比为1 ∶2, 改变AOT的含量以合成纳米SnO2粉末. 所合成粉末的XRD图谱如图2所示. 由图2知, 所获产物粉末均为金红石四方结构的SnO2. 以谢乐公式计算不同AOT含量下制得粉末的平均晶粒大小, 结果如表2所示. 由表2知, 当AOT含量为0.6 g时所制备产物晶粒最为细小, 说明此时表面活性剂包覆“水核”效果最好, 其产率也最高, 晶化程度适当. 当AOT含量低时, “水核”不能被表面活性剂完全包覆, 工艺过程中颗粒之间易产生“粘结”, 使得其晶粒粗大, 相应谱峰变得尖锐, 且产率降低. 当AOT含量为0.9 g时其基团数值增加, 使得微乳液的“水核”包裹过度, 晶粒虽然细小, 但氧化锡的产率明显降低, 且晶化程度不足. 因此, 选取AOT含量为0.6 g为最佳的表面活性剂含量.
2.2.2 热处理温度的影响
通过热分析可知, 600 ℃锻烧即可得到晶化较好的SnO2颗粒. 为进一步验证, 选用AOT含量为0.6 g, 保持其他条件不变, 热处理温度分别为500、 600、 700 ℃, 分别合成纳米SnO2粉末, 其XRD图谱如图3所示. 由图3可知, 随着煅烧温度升高, 半高宽变窄, 峰形变得愈加尖锐, 说明产物的晶化程度随之提高, 晶粒粒径变大. 以谢乐公式计算可得不同煅烧温度下制得SnO2粉末的平均晶粒大小, 分别为3.8 nm(500 ℃)、 4.1 nm(600 ℃)和11.9 nm(700 ℃). 煅烧温度在500 ℃时晶粒虽然细小, 但晶化不完全, 随后由500 ℃上升至600 ℃过程中, 晶粒长大不明显, 这与热分析研究的结果相一致. 分析其原因, 在此温度下颗粒表面包裹的有机物“壳”大都挥发殆尽, 对颗粒长大的阻碍作用基本消失, 故此现象或许与有机物改变了颗粒之间的界面张力有关. 当温度升高至600 ℃以上, 晶粒迅速长大, 这是由于此温度下有机物基本挥发分解完全, 颗粒之间不再有屏障, 随即发生剧烈团聚长大. 结合产率计算结果, 验证了热分析以600 ℃为最佳煅烧温度的结论.
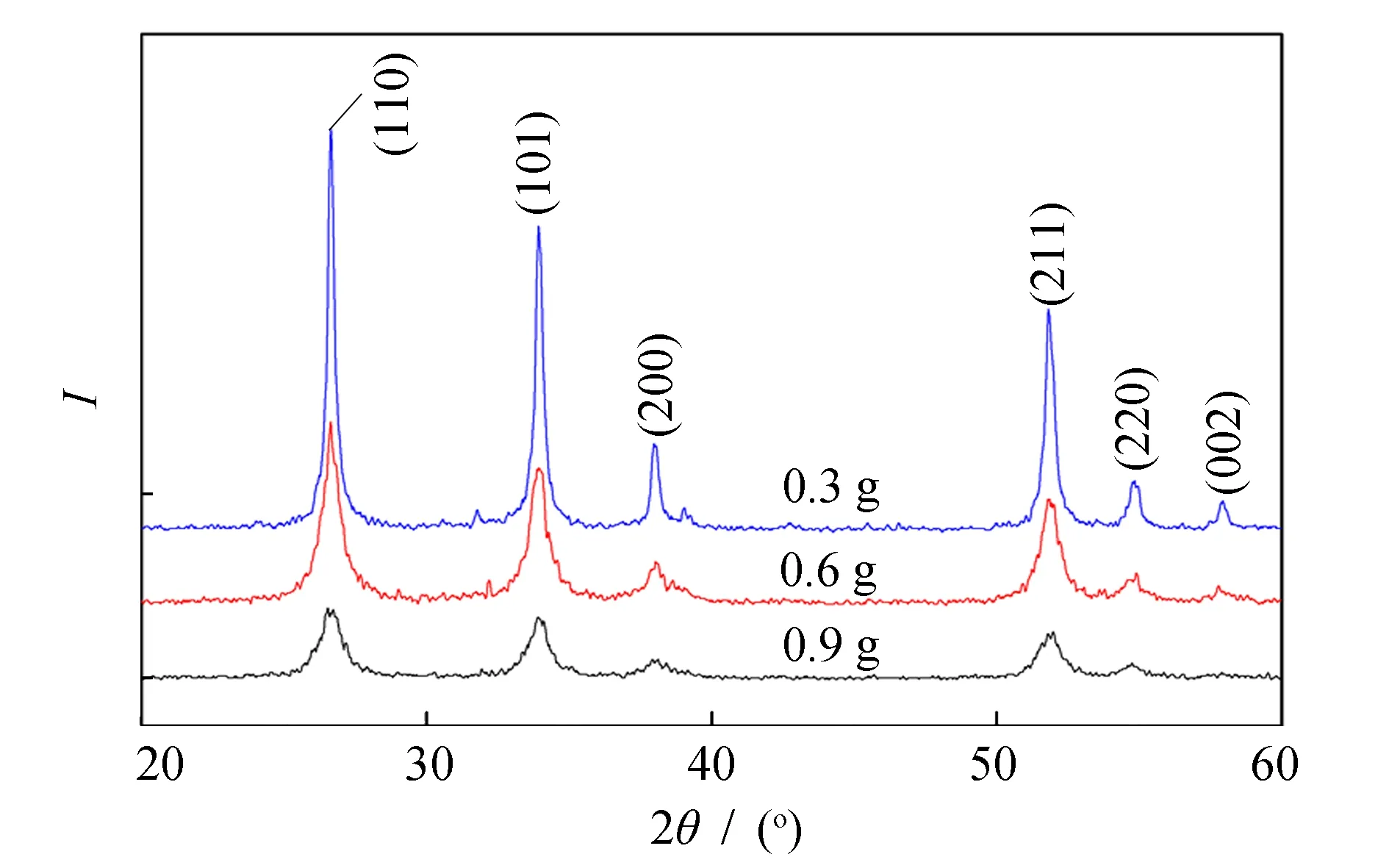
图2 不同AOT含量制备SnO2粉末 的XRD图谱(600 ℃, 2 h)Fig.2 XRD patterns of SnO2 powders calcined at 600 ℃ for 2 h prepared at different AOT contents

图3 不同煅烧温度下制备SnO2粉末 的XRD谱图(保温2 h)Fig.3 XRD patterns of SnO2 powders calcined at different temperatures for 2 h
2.2.3 溶液pH值的影响
选择AOT含量为0.6 g, 煅烧温度为600 ℃, 保持其他条件不变, 在溶液中加稀盐酸或氨水调整pH合成纳米SnO2粉末, 其XRD图谱如图4所示.
由图4知, 随着溶液pH值的升高, 谱图峰形变化明显, pH值为10和6时晶粒都较pH为8时更加粗大. 一方面, pH值过高, 导致已经形成的Sn(OH)2或者Sn(OH)4溶解, SnO2产率降低, 如图5所示. pH值降低为6时, 除去晶粒更粗大之外, 溶液中过多的H+抑制Sn4+与OH-反应, 将导致沉淀的减少, 产率随之下降. 由上述分析, 溶液pH值选在8左右为佳.

图4 不同pH值制备SnO2的 XRD图谱(600 ℃,2 h)Fig.4 XRD patterns of the SnO2 powders calcined at 600 ℃ for 2 h prepared at different pH value

图5 溶液pH值与SnO2产率的关系Fig.5 Dependence of pH value of the solution on rate of production of SnO2
2.3 红外分析
为研究煅烧前后产物分子结构的变化, 选取90 ℃干燥后粉末和600 ℃煅烧2 h的粉末分别进行红外测试, 图谱如图6所示. 由图6知, 波数在3 420~3 440 cm-1左右吸收峰为样品中吸附水O—H键的伸缩振动峰, 此峰在90 ℃干燥后仍十分明显, 说明样品中的吸附水量相当多, 这与其晶粒非常细小有关, 而当加热到600 ℃煅烧后, 此峰便明显减弱, 说明吸附水已经大量蒸发, 这与热分析的结果一致. 3 125、 2 875与2 950 cm-1左右的吸收峰均与Sn—OH键的振动有关, 此峰同样在600 ℃煅烧后基本消失, 证明在此温度下反应的沉淀物Sn(OH)4已经分解完全. 1 750 cm-1处的吸收峰为前驱物中AOT分子中羰基(C=O)伸缩振动, 但由于AOT分子中的羰基Sn4+络合配位而使得此吸收峰较AOT单分子的羰基吸收峰向长波方向移动了约200 cm-1, 即发生了红移. 1 667 cm-1处归属于水分子中O—H的变形振动. 1 385 cm-1左右吸收峰为晶格振动所致. 1 250 cm-1处的吸收峰对应于-CH2扭曲振动, 600 ℃煅烧后此峰消失, 有机物彻底分解. 1 150 cm-1处的吸收峰为-SO3中的S=O键的对称伸缩振动. 600 cm-1附近为O—Sn—O变形振动和Sn—O键伸缩振动导致的吸收峰. 整体谱图与文献[14-15]报道基本一致, 说明600 ℃煅烧可以使得产物完全转化为氧化锡, 有机物彻底分解. 与纯SnO2煅烧相比较[15], 谱图中增加了有机物的吸收峰, 此为微乳液法制备纳米颗粒的特征之一.
2.4 两种制备方法的比较

图7 CPM法和WOM法制备SnO2粉末的 XRD谱图(600 ℃) Fig.7 XRD patterns of the SnO2 powders calcined at 600 ℃ prepared by WOM and CPM methods
综上知, 采用微乳液法(WOM)制备SnO2纳米粉的最佳工艺是: 锻烧温度600 ℃、 保温时间2 h、 pH8、 水-表面活性剂摩尔比1 ∶2、 AOT含量0.6 g. 而采用化学沉淀法(CPM)的最佳工艺是: 锻烧温度600 ℃、 保温时间0.5 h、 pH5. 将最佳工艺条件下制备的SnO2粉末的XRD谱图加以对比, 如图7所示. 两种方法所制备的产物均为金红石结构的SnO2, 采用WOM所制备产物的谱峰较宽, 其中(111)晶面的峰尚未独立显现, 而采用CPM制备的SnO2图谱, 尽管煅烧时间只有前者的1/4, 但整体谱峰却已经变得更加尖锐, 其(111)晶面已经完全独立显现出来, 故可以定性推断, 同等加热条件下WOM所获晶粒较后者要精细. 采用谢乐公式计算后, 获得的平均晶粒度分别为: WOM, 4.1 nm; CPM, 10.1 nm,
前者所获晶粒的确更加细小. 考虑到除了粒子大小, 实际应用中另一个重要方面是其分散度, 故将两种方法所制备的SnO2粉末做了SEM表征, 结果如图8所示.由图8(a)知, 经600 ℃煅烧2 h后, 以微乳液法制备的纳米SnO2粉体主要表现为不规则多边形颗粒, 形状近似球体, 在2 500倍的放大倍数下颗粒总体仍然分布均匀, 团聚较少. 而图8(b)中, 同样是600 ℃煅烧, 虽然保温时间只有短短0.5 h, 但颗粒已经剧烈团聚, 在放大倍数为1 000倍下, 团聚的颗粒完全板结成致密的板块, 导致比表面积明显降低, 其性能也随之变差. 根据微乳液法的特点, 化学反应是在“水核”中进行的, 纳米粒子的成核、 生长过程都是在“水核”里完成. 正是由于这些表面活性剂分子包覆层的存在, 能有效阻止纳米粒子间的团聚, 使生成的纳米粒子之间相互间隔开来, 因此实现了纳米粒子的高分散特性. 故采用微乳液法制备氧化锡粒子不但可以通过调整溶液不同反应物的浓度来控制纳米粒子的大小, 更可以防止其团聚. 与沉淀法相比较, 具有粒径大小可控、 粒度分布均匀、 实验装置简单、 条件温和及操作方便等优点.

图8 不同方法制备SnO2纳米粉末的显微形貌Fig.8 Morphology of SnO2 calcined by different preparation method
3 结语
分别采用以AOT为添加剂的微乳液法和化学沉淀法制备了SnO2纳米粉体颗粒. 结果表明, 在600 ℃煅烧2 h后, 采用微乳液法制备的产物具有平均晶粒直径为4.1 nm的细小颗粒. 同样在600 ℃下煅烧, 采用化学沉淀法制备的粉末, 其保温时间减少为0.5 h, 但其平均直径已经长大到10.1 nm. 另外, 前者具有更加均匀的分散度. 说明采用以AOT为表面活性剂的微乳液法是合成细小分散SnO2纳米粉体材料的有效办法, 在AOT-正庚烷-SnCl4体系中, 制备SnO2纳米粉末的最佳工艺条件为: 锻烧温度600 ℃、 保温时间2 h、 pH=8、 水-表面活性剂摩尔比1 ∶2、 AOT含量0.6 g.
[1] Lee J H, Park N G, Shin Y J. Nano-grain SnO2electrodes for high conversion efficiency SnO2-DSSC[J]. Solar Energy Material and Solar Cells, 2011, 95(1): 179-183.
[2] Ansari S G, Boroojerdian P, Sainkar S R,etal. Grain size effects on H2gas sensitivity of thick film resistor using SnO2nanoparticles[J]. Thin Solid Films, 1997, 295(1/2): 271-276.
[3] Gu Feng, Wang Shufen, Song Chunfeng,etal. Synthesis and luminescence properties oF SnO2nanoparticles[J]. Chemical Physics Letters, 2003, 372(3/4): 451-454.
[4] Batal M A, Jneed F H. Tin oxide n-type semiconductor inverted to p-type semicangductor prepared by sol-lel method[J]. Energy Procedia, 2011, 6(1): 1-10.
[5] Hou Linrui, Yuan Changzhou, Peng Yang. Synthesis and photocatalytic property of SnO2/TiO2nanotubes composites[J]. J Hazard Mater, 2007, 139(2): 310-315.
[6] Cho Y S, Yi G R, Hong J J,etal. Colloidal indium tin oxide nanoparticles for transparent and conductive films[J]. Thin Solid Films, 2006, 515(4/5): 1 864-1 871.
[7] Cachet H, Campet A G, Jousseaume B G,etal. Tin dioxide thin films prepared from a new alkoxyfluorotin complex including a covalent Sn-F bond[J]. Thin Solid Films, 2001, 388(1/2): 41-49.
[8] Norris D J, Efros A L, Erwin S C. Doped nanocrystals[J]. Science, 2008, 319(5 871): 1 776-1 779.
[9] Malik M A, Wani M Y, Hashim M A. Microemulsion method: a novel route to synthesize organic and inorganic nanomaterials[J]. Arabian Journal of Chemistry, 2012, 5(4): 397-417.
[10] Song K C, Kim J H. Preparation of nanosize tin oxide particles from water-in-oil microemulsions[J]. Journal of Colloid and Interface Science, 1999, 212(1): 193-196.
[11] Chen Deliang, Gao Lian. Novel synthesis of well-dispersed crystalline SnO2nanoparticles by water-in-oil microemulsion-assisted hydrothermal process[J]. J Colloid Interface Sci, 2004, 279(1): 137-142.
[12] 霍涌前, 王升文, 程丽丽, 等. SnO2的微乳液法合成及其气敏性能测试[J]. 延安大学学报: 自然科学版, 2011, 30(1): 58-61.
[13] Luwang M N, Ningthoujam R S, Singh N S,etal. Surface chemistry of surfactant AOT-stabilized SnO2nanoparticles and effect of temperature[J]. Journal of Colloid Interface Sci, 2010, 349(1): 27-33.
[14] Amalric-Popescu D, Bozon-Verduraz F. Infrared studies on SnO2and Pd-SnO2[J]. Catalysis Today, 2001, 70(1/2/3): 139-154.
[15] 孙明, 余林, 郝志峰, 等. SnO2纳米粒子的制备与表征[J]. 无机化学学报, 2005, 21(6): 925-928.
(责任编辑: 郑美莺)
Preparation and characterization of nanosize SnO2particles with AOT surfactant from water-in-oil micromulsions
LIU Xuehua1, 2, 3, TANG Dian2, 3
(1.School of Materials Science and Engineering, Fujian University of Technology, Fuzhou, Fujian 350118, China; 2. Fujian Province Key Laboratory of Advanced Materials Processing and Application, Fuzhou, Fujian 350116, China;3.College of Materials Science and Engineering, Fuzhou University, Fuzhou, Fujian 350116, China)
Nanoparticles of tin oxide (SnO2) have been prepared from water-in-oil (W/O) micro-emulsions consisting of SnCl4, water, AOT, and n-heptane. The effects of calcination temperature, AOT contents and pH values of solution on the properties of products were studied by XRD, DTA-TG, FTIR and SEM. Results show that the optimal condition in the system is: pH value of the solution was 8, the molar ratio of water to AOT was 1 ∶2, the content of AOT was 0.6 g and the products was calcined at 600 ℃ for 2 h. The tin oxide powder was found to be less than 4.1 nm in particle diameter and to be more disperse than it prepared by the chemical precipitation method.
AOT; microemulsion; SnO2; nanosize; preparation; characterization
10.7631/issn.1000-2243.2015.04.0542
1000-2243(2015)04-0542-06
2014-05-29
刘雪华 ( 1975-), 副教授, 主要从事纳米功能材料研究, paopaotu326@163.com
国家自然科学基金资助项目(11374053); 福建省教育厅科研资助项目(JA12233)
O 647.11; TQ 610.494
A
猜你喜欢
环境卫生工程(2021年5期)2021-11-20 05:45:30
当代化工研究(2016年2期)2016-03-20 16:21:20
中国果菜(2016年9期)2016-03-01 01:28:39
中国造纸(2015年7期)2015-12-16 12:40:48
化工进展(2015年6期)2015-11-13 00:32:12
中国洗涤用品工业(2015年11期)2015-02-28 19:03:09
中国洗涤用品工业(2015年2期)2015-02-28 19:01:57
中国洗涤用品工业(2015年2期)2015-02-28 19:01:56
济宁医学院学报(2014年4期)2014-08-16 13:44:19
无机化学学报(2014年5期)2014-02-28 17:31:32
