
Enhancement of solubility and therapeutic potential of poorly soluble lovastatin by SMEDDS formulation adsorbed on directly compressed spray dried magnesium aluminometasilicate liquid loadable tablets:A study in diet induced hyperlipidemic rabbits
2015-05-15 08:29:24MohdJvedQureshiChitneniMllikrjunWongGnKin
Mohd Jved Qureshi,Chitneni Mllikrjun,Wong Gn Kin
aDepartment of Pharmaceutical Technology,School of Pharmacy,Taylors University,Lakeside Campus,Selangor, Malaysia
bDepartment of Pharmaceutical Technology,School of Pharmacy,International Medical University,Kuala Lumpur, Malaysia
Enhancement of solubility and therapeutic potential of poorly soluble lovastatin by SMEDDS formulation adsorbed on directly compressed spray dried magnesium aluminometasilicate liquid loadable tablets:A study in diet induced hyperlipidemic rabbits
Mohd Javed Qureshia,b,*,Chitneni Mallikarjuna,Wong Gan Kiana
aDepartment of Pharmaceutical Technology,School of Pharmacy,Taylors University,Lakeside Campus,Selangor, Malaysia
bDepartment of Pharmaceutical Technology,School of Pharmacy,International Medical University,Kuala Lumpur, Malaysia
ARTICLEINFO
Article history:
Received 28 April 2014
Received in revised form
17 July 2014
Accepted 3 August 2014
Available online 2 September 2014
Lovastatin
Self-microemulsifying drug delivery
system(SMEDDS)
Neusilin®US2
Liquid loadable tablet
Solid carrier system
Pharmacodynamics studies
The aim of present study was to formulate and evaluate a self-microemulsifying drug delivery systems(SMEDDS)containing lovastatin and to further explore the ability of porous Neusilin®US2 tablet as a solid carrier for SMEDDS.SMEDDS formulations of varying proportions of peceol,cremophor RH 40 and transcutol-P were selected and subjected to invitro evaluation,including dispersibility studies,droplet size,zeta potential measurement and release studies.The results indicated that the drug release prof i le of lovastatin from SMEDDS formulations was statistically signif i cantly higher(p-value<0.05)than the plain lovastatin powder.Thermodynamic stability studies also conf i rmed the stability of the prepared SMEDDS formulations.The optimized formulation,which consists of 12%of peceol,44%of cremophor RH 40,and 44%of transcutol-P was loaded into directly compressed liquid loadable tablet of Neusilin®US2 by simple adsorption method.In order to determine the ability of Neusilin®US2 as a suitable carrier pharmacodynamics study were also carried out in healthy diet induced hyperlipidemic rabbits.Animals were administered with both liquid SMEDDS and solid SMEDDS as well.From the results obtained,Neusilin®was found to be a suitable carrier for SMEDDS and was equally effective in reducing the elevated lipid prof i le.In conclusion,liquid loadable tablet(LLT)is predicted to be a promising technique to deliver a liquid formulation in solid state.
©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
1.Introduction
Currently,majority of the new drug molecules being discovered are lipophilic and exhibits poor water solubility which results in low bioavailability,intra and inter subject variation and lack of dose proportionality[1].Limited water solubility posses a challenge in developing optimum oral solid dosage form in terms of formulation design,bioavailability and marketing of new pharmaceutical products.Several formulation strategies have been approved to overcome these challenges eitherbymeansofmodifyingthesolubilizationormaintaining the drug in dissolved form throughout gastric transit time [2-4].These strategies may include the use of surfactants, cyclodextrins,micronization,salt formation,pH change,nano size delivery,solid dispersions and permeation enhancers [1,5].Infact,mostcommonlyusedapproachesare,digestionof the active pharmaceutical ingredient into inert lipids such as oils and surfactant dispersions[6],self-emulsifying formulations[7-9]emulsions[10]and liposomes[11,12].
SMEDDS,an emulsion based formulation is a blend of oils and surfactants in suitable proportion that rapidly forms an oil in water(o/w)microemulsion with moderate gastric motility when exposed to the aqueous media present in the g.i.t[13].Co-surfactant and organic solvent can also be added sometimetoimprovetheemulsif i cationandsolubility respectively.SMEDDS typically produce a transparent microemulsion having droplet size of<50 nm[14]and are physically and thermodynamically stable formulations.Rapid emulsion formation helps to keep the drug in a dissolved form,however,small droplet size offers a considerably larger interfacial surface area which further accelerate the absorption rate of drug with limited solubility.This feature makes SMEDDS a meaningfulchoicefororaldeliveryoflipophilic,low bioavailable drugs having adequate lipid solubility[13,15-17].
Moreover,the lipoidal part of SMEDDS encourages the intestinal lymphatic uptake of drugs which further helps in avoiding the presystemic biotransformation of drug molecules.Lovastatin undergoes substantial f i rst-pass metabolism facilitated by CYT P450 3A4 in liver.The basic mechanism in improving the bioavailability by SMEDDS comprises of increaseinthemembranef l uidityduetopresenceofsurfactants and co-surfactant which expedite transcellular absorption [18-22].Surfactant and co-surfactant molecules get favorably absorbedattheliquidinterfaceduringtheprocessofemulsion formation,reducing the interfacial energy of the system.This helps spontaneous emulsif i cation without high energy input. Thus,well-designed SMEDDS formulation can ensure eff i cient self-emulsif i cation as well as greater solubilization capability for the drug in the resultant dispersion[23].
Choosing the right combination of lipid(oil),surfactant, andco-surfactantisoneoftheimportantpointsin designing SMEDDS formulations.Selection of a good selfmicroemulsifying formulation depends on the(1)the solubility of the drug in oil/surfactant/co-surfactant(2)emulsion forming area as determined by phase diagram,and(3)the globule size distribution of the developed SMEDDS[24,25].
There are various reasons or challenges in developing lipid based systems,like so far there are no def i nite in-vitro evaluation or characterization tests which can simulate or predict the in-vivo performance of drug administered in lipoidal formulations[26,27],second is of course liquid nature of these formulations which make them failure at commercial success because of diff i culties in manufacture,handling,storage, supply and same time retain risk of stability issues too. Because of these reasons SMEDDS formulation can only be supplied either bottle f i lled or if dose is low then it could be supplied in liquid f i lled soft gelatin capsule as well.Such kind of formulations have many restrictions,f i rst of all oral solution are not very well accepted by the patient and second capsule f i lled formulation have dose limit.Drug with higher doseis diff i cultto incorporatein limited spaceavailable in soft gelatin capsule shells.Furthermore there is a risk of leaching and precipitation if capsules shells are not properly sealed or manufactured[28].
To overcome these limitations of liquid SMEDDS many different approaches like spray drying[29],melt granulation, adsorption on solid carrier[30-32]and other techniques[33] have been explored to transform the lipid based formulation into solid oral dosage form.Conversion of liquid formulations into a suitable solid dosage form is an exciting but diff i cult task too.From many years soft or hard gelatin capsules are widely being used to f i ll and administer the liquid dosage form.However,these capsules are also accompanying various formulation related problems like leaching of material,leaking,sticking,physical incompatibilities with capsule shell, slower production rate compared to tablet etc.Tablets are apparently a better substitute to liquid f i lled soft gelatin capsule to administer SMEDDS in terms of patient compliance and physical stability.Moreover,tablet can take a higher drug load compared to capsule formulation.The major constraint in transformation of SMEDDS into a tablet form is availability of an appropriate carriermolecule whichcan take up a desired quantity of liquid formulation and at the same time can exhibit a good tablet quality as well.Liquid formulation can easily be converted into free f l owing powder form by adsorbing on suitable solid carrier particles like silicate derivatives, dextran,carbon tubes[33]and silicon dioxide.Subsequent to adsorption these lipid loaded solid carrier can either be f i lled directly into capsule housing or can be mixed with other excipient required for tablet compression.Although this carrier approach seems to be effective and easy but there are certain constraints or limitations accompanied with this approach such as,when a solid carrier loaded with high liquid content is subjected to compression,adsorbed liquid component may exudate out and problems of chipping,sticking, variable hardness and soft tablets are likely to be present. Second,a large quantityofsolid carrierisneededto adsorbthe liquid formulation which eventually will lead to large volume of f i nal dosage form[34].
The present study is focused to overcome these shortcomings of tablet compression by exploring the use of Neusilin®US2 tablets as solid carrier followed by adsorption of liquid SMEDDS formulation.Neusilin US2(Fuji chemicals,JP) is a spray dried synthetic amorphous form of magnesium aluminometasilicate(MAMS)having large surface area and high oil and water adsorption ability.MAMS is completely insoluble in water but have slight solubility for acid and alkali [35].Lovastatin was chosen as the drug to be incorporated intothe SMEDDS formulation,due to its low aqueous solubility. Lovastatin is a cholesterol-lowering agent which acts by inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A(HMGCoA)reductase,which accelerates the translation of HMG-Co enzyme A to mevalonate which serves as precursor to cholesterol in the biosynthetic pathway of cholesterol.Lovastatin is reported to have less than 5%bioavailability of an orally administered dose due to extensive f i rst-pass metabolism[36].It is classif i ed as a BCS Class II,with“low solubility/high permeability”therefore,it can be anticipated that the poor oral bioavailability of lovastatin could be due to its limited aqueous solubilization which further poses dissolution limitations[37].
The aim of present study was to develop an appropriate SMEDDS formulation for lovastatin to improve its bioavailability and also to explore the capability of LLT made from MAMS as a carrier to be loaded with self-microemulsifying formulation in order to enhance the physical and chemical stability of developed delivery system.Finally the developed lovastatin-SMEDDS and LLT loaded with SMEDDS containing lovastatin were evaluated for pharmacodynamics eff i ciency in diet induced hyperlipidemic rabbits.
2.Material and methods
2.1.Materials
Lovastatin(Shanghai PI Chemicals Ltd(China)),Neusilin®US2(Fuji Chemical Japan),Crospovidone(Polyplasdone XL 10, ISP Technologies,USA),Polyglycolysed glycerides(Capryol 90, Capyrol PGMC,Lauroglycol 90,Labrasol,Transcutol-P,Labraf i l M 1944 CS,Labrafac PG,Labrafac Lipophile and Peceol)were provided as gift samples by Gattefosse(France).Cremophor RH 40 was provided as gift samples by BASF Germany.Tween 80,Tween 20,Tween 40,Tween 60,Span 80,Span 20,polyethylene glycol(PEG)300,PEG 400 and PEG 600,magnesium stearate were purchased from Sigma-Aldrich(USA),Methanol of HPLC grade was purchased from J.T.Baker(USA).All the other chemicals and reagents used were of analytical grade.
2.2.Methods
2.2.1.Solubility studies
In order to select a best combination of oils,surfactants,and co-surfactants for SMEDDS formulation,component which shown a maximum solubility for lovastatin was selected.An excess quantity of lovastatin was taken into each screwcapped glass vials which contains 2 ml of the respected vehicle.Vials were hermetically sealed and warmed at 40°C in a water bath followed by stirring on a vortex mixer to facilitate the solubilization.Later,samples were kept at room temperature for 72 h in shaker bath of 150 oscillations/min.After reachingequilibrium,thesupersaturatedmixturewas centrifuged at 3000 rpm for 15 min and undissolved lovastatin was separated by f i ltration thru 0.45 μm pore size f i lter[24,38]. The amount of solubilized lovastatin was quantif i ed by the HPLC analysis.The results were treated statistically using one-way analysis of variance(ANOVA),in order to determine the statistically signif i cant differences among the solubility of the vehicles in each category.
2.2.2.Pseudo-ternary phase diagram
As per the solubility data presented in Table 1 cremophor, transcutol-P and peceol,were chosen as surfactant,cosurfactant and the oil phase respectively.To determine the concentration of components for the prevailing range of SMEDDS,pseudo-ternary phase diagram was drawn using water-titration method at ambient temperature[39].Different surfactant/co-surfactant mixtures(Smix)were prepared by mixing theselectedsurfactant andco-surfactant at threef i xed ratios(1:1,2:1 and 3:1 w/w).The selected oil was then added to each Smix at eight different ratios(9:1,8:2,7:3,6:4,5:5,4:6,3:7, 2:8 and 1:9 w/w).Water was incorporated to each mixture in 5%step-wise increments[40]followed by gentle swirling of the mixture.After every water addition in dropwise manner, the mixture was visually observed for its transparency and turbidity.The end point of turbidity-to-transparency and transparency-to-turbidity transitions was noted[24],and the percentages of water,oil and Smix were calculated at this point.Based on these percentages phase diagrams were plotted,and the microemulsion region in each phase diagram was identif i ed.In order to observe the inf l uence of drug addition on the microemulsion forming area,phase diagrams were also constructed with drug(lovastatin)using drug-oil mixture as the hydrophobic component[41].
2.2.3.Preparation of SMEDDS formulation
A range of formulation was formulated using transcutol-P, cremophor RH 40 and peceol as co-surfactant,surfactant and oil phase respectively.Lovastatin(10 mg/ml)was added into the oily phase in small increment with continuous stirring. The surfactant system(Smix)was prepared separately by mixing the selected surfactant and co-surfactant in their determined ratios chosen from the microemulsion region of the constructed phase diagram[42].Oil phase containinglovastatinwasaddedintothesurfactantsystemwith continuous stirring and vortex mixing till the homogenous mixture was formed.The resulting microemulsions were stored for a day and inspected for any possibility of physical instability characterized by phase separation and drug precipitation.Furthermore,in order to select an optimized SMEDDS formulation,the effect of oil to surfactant+co-surfactant(O:Smix)ratio,susceptibility of droplet size for change in pH of the dilution medium were also taken into consideration[18].
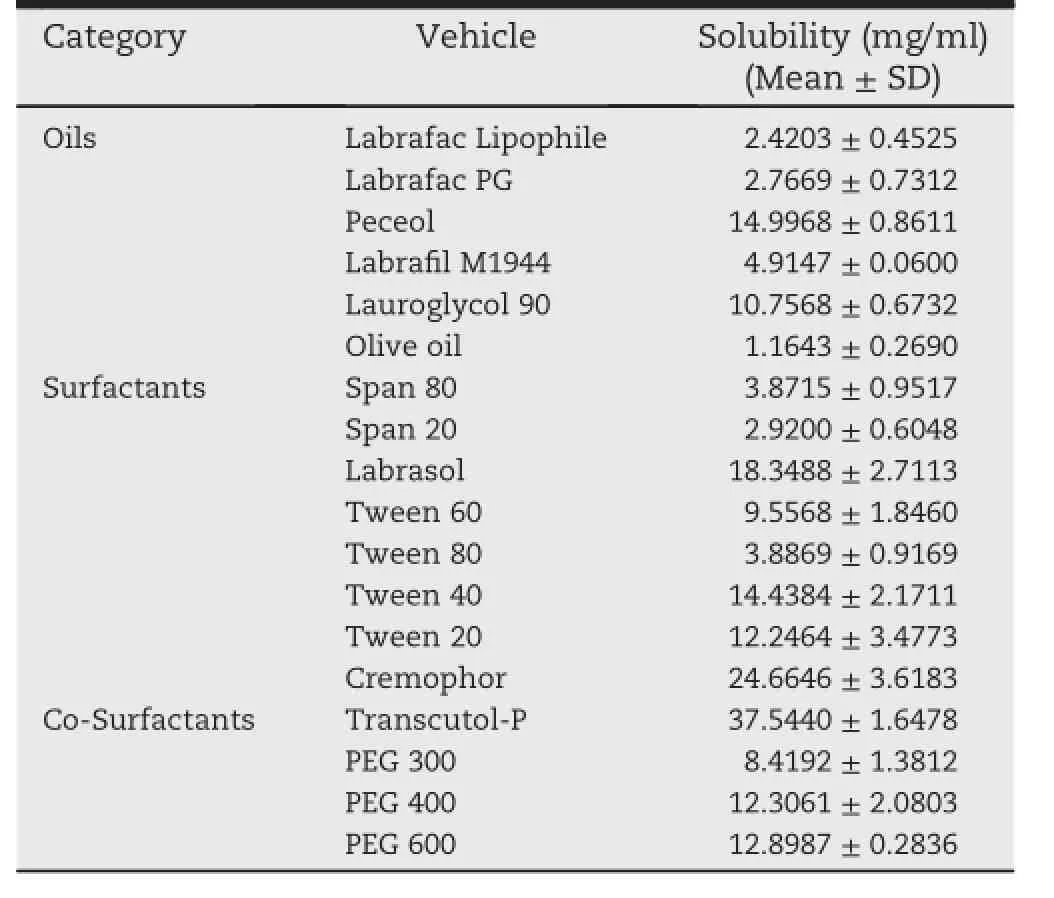
Table 1-The solubility of lovastatin in different oils, surfactants and co-surfactants.
2.2.4.Thermodynamic stability study
These studies,aimed at evaluating the stability of the formulations prepared,consist of two phases.Phase I is the heating cooling cycle,where all the formulations were subjected to six cycles between 4°C and 45°C,with storage at each temperature of minimum of 48 h.After complete cycles of alternate temperatures,formulations were centrifuged at 3500 rpm for 15 min to observe for any phase separation or drug precipitation.Formulations which succeeded in phase I were taken to phase II(freeze-thaw cycle),where the formulations were subjected to three cycles of alternate temperature between-21°C and+25°C.Samples were stored for minimum of 48 h at each temperature[42].Succeeding to phase II,in order to determine the physical stability,formulations were further allowed to centrifuge at a speed of 3500 rpm for 15 min for possibility of phase separation or drug precipitation.Formulations which were found to be stable at alternate temperature cycles were selected for further evaluation.
2.2.5.Globule(droplet)size and zeta potential analysis
1 ml of the formulation was diluted to 10 ml using distilled water.An aliquot of the resultant emulsion was then subjected to the Malvern Zetasizer for droplet size and zeta potential measurement at pH 7.0 and 25°C.
2.2.6.Assessment of self-emulsif i cation
In order to determine the eff i ciency of self-emulsif i cation of various mixtures,1 ml of each formulation was added to 250 ml of distilled water contained in a vessel of USP apparatus 2,with gentle agitation,provided by a paddle rotating at a speed of 50 rpm.Apparatus was maintained at a temperature of 37°C.Self-emulsif i cation process was visually monitored for the emulsif i cation rate and for the quality of the produced microemulsion using grading system give in Table 4.
2.2.7.Precipitation analysis
The prepared SMEDDS were diluted with 0.1 N HCl up to 250 times.The diluted microemulsion was carefully examined for any indication of phase separation or drug precipitation at time period of 1st hour and followed by at 6th hour[43].
2.2.8.In-vitro drug release study of lovastatin-SMEDDS
In-vitrodrugreleasestudiesfor preparedSMEDDSwerecarried out in 0.1 N HCl and pH 6.8 phosphate buffer solution using Tablet Dissolution Test Apparatus,Type II(paddle method)at 37°C±0.5°C and at paddle speed of 50 rpm.A quantity of SMEDDS formulations equivalent to 10 mg of lovastatin was taken into vessels containing 900 ml of dissolution media. Throughout the release studies,an aliquot of 5-ml was withdrawnatpredeterminedtimeinterval,f i lteredthrough membrane f i lter and analyzed using UV/Spectrophotometer at 240 nm.Subsequently withdrawn sample was replaced with equal volume of fresh buffer solution to compensate for the loss due to sampling and to maintain the sink condition [25].The dissolution eff i ciency at 15th minute(DE15min)for the formulations and plain drug powder were also determined and compared.
2.2.9.Preparation of liquid loadable tablet(LLT)
LLT was prepared by direct compression method and is comprised of MAMS/Crosspovidone XL 10/magnesium stearate/Talc(88%/10%/1%/1%)in a given proportions.All excipients passed through sieve 180 μm mesh size to break any lumps or aggregates,were loaded into lab scale conta blender (Mec well Pharma Machinery company,Thane,Mumbai, India)and mixed thoroughly prior to compression.Following to mixing,blend was directly compressed using round,10 mm fl at punch tooling.Tablets were evaluated for hardness, disintegration and dissolution behavior.
2.2.10.Loading of SMEDDS into LLT
Before loading of liquid formulation,compressed LLT were evaluated for porosity and loading capacity(ml).LLT was considered fully loaded when the theoretical limit is achieved which implicates that void spaces are completely saturated with liquid formulation.In order to load the LLT, tablet were placed in contact with excess of optimized lovastatin-SMEDDS formulation and allowed to adsorb the liquid until a constant weight of tablet is achieved.Prior to weigh,excess of liquid present over the tablet was wiped off and dried with a tissue paper.Fully loaded tablets of constant weight were stored in air tight container for further use.
Tablet porosity:Tablet porosity was determined using following formula.
where ρpand ρtrepresents the pycnometric tablet density and tablet density respectively.
2.2.11.In-vitro release behavior of loaded LLT
SMEDDS loaded tablets were subjected to in-vitro dissolution studies in purif i ed water using USP apparatus type 1(basket type)set at paddle speed of 50 rpm and temperature of 37°C.
2.2.12.Shelf life determination of the optimized formulations Accelerated stability studies were carried out to determinethe storage life of developed formulation.The SMEDDS formulation were stored at 30°C,40°C and 50°C at normal room humidity conditions for 60 days.Samples were withdrawn from each temperature condition after predetermined time period(0,15,30,45 and 60 days)and analyzed for drug content using the HPLC method[42].The order of the rate of degradation reaction and the degradation rate constant wascalculated from the slope of lines at each temperature using the below equation:
The plot of Log K vs.1/T at various temperatures was drawn.From the plot K value corresponding to 25°C was calculated and was used to determine the shelf life of formulation using following formula[44].
where t0.9is the time required for 10%degradation of the drug and is represents the shelf life[45].
2.2.13.Pharmacodynamics effect of lovastatin-SMEDDS and lovastatin-SMEDDS loaded tablets
Eighteen healthy male New Zealand white rabbits weighing 1.5-2.0 kg were purchased from East Asia Rabbit Corporation Sdn Bhd Malaysia and kept indoors in individual cages Animal room was maintained under constant environmental conditions(22°C±2°C,40-70%RH)and had alternating 12 h dark andlightcycle[46].Allanimalswereacclimatizedintheanimal roomfortwoweekspriortoexperimentsandduringthisperiod they had free excess to normal Ralston Purina®laboratory RabbitchowComplete(PurinaMills,St.LouisMissouri)pelleted diet and drinking water ad libitum.Animal handling and all the experimental work was done according to protocol and standard operating procedure(SOPs)approved by international medical university research and ethical committee.After 2 weeks of acclimatization,Overnight fasted animals were divided into 6 groups namely G1-G6(3 Rabbits in one group). Control group(G1)with normal cholesterol level was maintainedonthestockdietofPurinaChow.Thisfeedismadefrom vegetable sources and contains 16%crude protein,1.5%crude fat,17%crude f i ber and vitaminA 4650IU/lb.Theremaining 16 rabbits(G2,G3,G4,G5 and G6)were subjected to experimental treatmentandarefedwith2%contentcholesterolPurinarabbit chow(Dytes Inc)for 3 weeks in order to induce acute hypercholesterolemia.Group 2 animals were maintained on high cholesteroldietwhereasGroup3wasfedwithhighcholesterol dietfollowedby placebo formulationandserved ascontrol i.e., with acute hypercholesterolemia but no drug,G4 was administered with lovastatin suspension(6 mg/kg),G5 was administered with lovastatin-SMEDDS(6 mg/kg)and f i nally the G6 was administered with LLT loaded with lovastatin-SMEDDS (6 mg/kg).The blood was withdrawn from marginal ear vein of rabbits at weekly interval and serum cholesterol,total lipid content was determined using in-vitro diagnostic kit (Cardiochek,San Diego,USA).Determination of serum HDL, LDL,triglyceride was carried out using Abcam's diagnostic kit according to the manufacturer's instructions.Abcam's HDL and LDL/VLDL Cholesterol quantif i cation assay kit offers a simple method for quantif i cation of above said components following a proper separation of HDL from LDL and VLDL(very low density lipoprotein)in serum samples[47].This is an enzymatic colorimetric test.In the assay,cholesterol oxidase distinguishes free cholesterol and develops products which interact withprobeto produce color(570nm)andf l uorescence (Ex/Em=538/587 nm).Kit was stored at-20°C.
2.2.13.1.High cholesterol diet(HCD).A 2%cholesterol diet was prepared by adding 2 g of pure cholesterol powder analytical grade(C75209,Sigma Chemical Co.,St.Louis,USA)to each 100 g of grounded rabbit chow pellet(2%cholesterol,w/w,in food pellet).Before mixing to rabbit chow pellets,cholesterol powder was dissolved in 40 ml of chloroform in order to achieve a uniform distribution throughout the feed.After thorough mixing Chloroform was evaporated by heating the diet in Hot air oven maintained at 50°C[48,49].
2.2.13.2.Separation of HDL and LDL/VLDL.100 μl of 2×precipitation buffer was mixed with 100 μl of serum sample in microcentrifuge tubes.Mixture was incubated for 10 min at room temperature,followed by centrifugation at 2000×g.The separated HDL supernatant was transferred into fresh tubes and labeled.Precipitates were consists of mixed fraction of LDL and VLDL.Results of testing were multiplied by time 2 to compensate the dilution with 2×precipitation buffer solution [50].
In order to separate the LDL/VLDL precipitate was spun again at 2000×g,traces of HDL present in supernatant was removed carefully.LDL/VLDL fraction was suspended in phosphate buffer pH 7.4 and transferred to fresh tubes and stored[50].
2.2.13.3.Preparation of standard curve and sample preparation for colorimetric assay.20 μl of cholesterol standard provided with the kit was mixed with 140 μl of cholesterol assay buffer to produce a stock concentration of 0.25 μg/μl.An aliquot of 0, 4,8,12,16,20 μl was added into series of 96 well-plates.Volume wasadjustedto50μl/wellwithcholesterolassaybufferin order to produce 0,1,2,3,4,5 μg/well of the cholesterol standard[50].
2.2.13.4.Preparation of reaction mix.Reaction mix was prepared by mixing cholesterol assay buffer,cholesterol probe, and enzyme mix and cholesterol esterase[47].
2.2.13.5.Assay.50 μl of reaction mix was added into well containing cholesterol standard or test samples,reaction was incubated for 60 min at 37°C,protected from light.Optical density was measured at 570 nm in colorimetric microtiter plate reader[50].
3.Results and discussion
One of the main factors to be considered when developing a self-emulsifying formulation is to escape of any potential drug precipitation upon its dilution in the gastric f l uid[42].Ideally SMEDDS formulation is a mixture of oil,surfactants,cosurfactants with drug and it should produce a transparent clear single phase liquid at normal conditions when mixed with aqueous phase[43]and should have good solubilizing capacity for drug under study.In order to have the drug in solution form[51],it is assumed that the ingredients used in the SMEDDS could enhance the solubility and permeability of drug by considerably reducing the droplet size[52].Determination of oil with the highest solubilization capacity for thedrug is particularly important,since the ability of the microemulsion formed upon dilution to retain the drug in solubilizedform islargely dependenton itssolubility in the oilphase ofthe system.If the surfactant andco-surfactant contribute to a great extent for the solubilization of the drug,there possess a risk of drug precipitation upon dilution with aqueous phase. In addition,the surfactant selected should be capable of lowering the surface tension to a very low level to aid the dispersion process.Generally,non-ionic surfactants with high HLB values are used in SMEDDS formulation,as it had been reported that surfactants with HLB value between 12 and 15 are considered to possess good eff i ciency for self-emulsif i cation[24].Apart from that,the incorporation of co-surfactants along with surfactant to the system also enables the dissolution of signif i cantly large amount of either the drug or hydrophilic surfactants in the hydrophobic(oil)phase[4]. Organic solvent like polyethylene glycol(PEG),propylene glycol(PG)and transcutol-P are particularly useful as cosurfactant in formulating SMEDDS,since they are suitable for oral delivery.
3.1.Solubility study
The solubility of lovastatin in the different oils,surfactants and co-surfactants is presented in Table 1.The results of oneway ANOVA showed that there are statistically signif i cant differences among the solubility of different oils,since the p-value(labeled“sig”)is less than 0.05.Peceol has shown the highest solubility among the oils.The results obtained of Post-Hoc Tukey test clearly revealed that there are statistically signif i cant differences between peceol and labrafac lipophile,labrafac PG.Llbraf i l M1944 as well as Olive Oil,with a p-value<0.05.Among all the surfactants and co-surfactant studied,cremophor and transcutol-P showed the highest solubility for lovastatin(24.66 mg/ml and 37.54 mg/ml respectively).A statistically signif i cant difference was found amongthesolubilityofdifferentsurfactantandcosurfactants(p-value<0.05).The results of Tukey Post-hoc test clearly indicates that there are statistically signif i cant difference between cremophor and all the other surfactants (p-value<0.05),except labrasol(p-value=0.062;>0.05)and Tween 40(p-value=0.2262;>0.05)as well as transcutol-P and with all other co-surfactants,including PEG 300,PEG 400 and PEG 600(p-value<0.05).As per the results obtained after solubility studies,peceol,cremophor and transcutol were chosen as oil,surfactant and co-surfactant respectively for the proposed SMEDDS formulation.
3.2.Construction of pseudo-ternary phase diagram
The purpose of constructing pseudo-ternary phase diagram is to determine the microemulsion area for the selection of appropriateconcentrationsofoil,surfactantandco-surfactant in subsequent SMEDDS formulation development.There are few factors which determine the range of microemulsion region formed during the water-titration,including the physical and chemical properties of the oil phase,aqueous phase and surfactants used[53,54]as well as the necessary conditions needed for microemulsion formation.To be precise,these required conditions would include a less interfacial tension at the hydrophilic and hydrophobic interface,the presence of highly f l uid and f l exible interfacial f i lm as well as the penetration and association of oil molecules with the interfacial surfactant f i lm[53,54].Hydrophilic-lipophilic balance(HLB) value of surfactant and co-surfactant used in formulation is a key factor for the development of self-emulsion.Surfactants having HLB in the range of 12-15 are generally considered to have good eff i ciency for self-emulsif i cation[53,55].
Phase diagram were constructed at three different ratios of surfactant to co-surfactant(1:1,2:1 and 3:1)as shown in Figs. 1-3.Experiment was repeated in triplicate as presented by A, B and C for each ratio.Among all three ratios,it is evident that the surfactant to co-surfactant ratio of 1:1 produced a comparatively largest microemulsion region than the other two ratios as shown in Fig.1A-C.Results are also summarized in Table 2.The one-way ANOVA suggests that there are statistically signif i cant differences among the mean microemulsion area at different ratio of surfactant:co-surfactant (1:1,2:1,3:1),as the p-value is less than 0.05(p-value=0.008). Since the phase diagram of the ratio 1:1 yielded the greatest area of microemulsion region,the procedure of water-titration was repeated in triplicate in the presence of lovastatin for the same ratio to explore the effect of drug addition on the boundaries of microemulsion region as shown in Fig.4A-C.
A decrease in the area of the microemulsion region was observed following incorporation of 10 mg lovastatin.The mean microemulsion area of the phase diagrams constructedwithlovastatinaddedwascalculatedtobe 39.57 cm2,which is smaller than the phase diagram constructed without lovastatin added at the same ratio(1:1).A decrease in the microemulsion area is attributed to the incorporation of the drug into the oil droplets(lipid phase) resulting in the swelling of the oil droplets or expansion of lipid phase.Hence,an increase in the droplet size of the emulsion formed leads to a decrease in the clear region (microemulsion area)in the pseudo-ternary phase diagram which in turn leads to a need for a higher S/CoS ratio for emulsion stability[43].
The results of paired sample T-test suggest that there is a statistically signif i cant difference between the mean microemulsion area of the phase diagram at ratio 1:1 constructed with and without lovastatin added.
3.3.Formulation of self-emulsifying microemulsion
A total of eight combinations were selected from the microemulsion region of the pseudo-ternary phase diagram constructed with added lovastatin.The percentages of peceol,cremophor and transcutol-P in each formulation were illustrated in Table 3.All the selected eight SMEDDSformulations passed the thermodynamic stability tests,as none of them showed any signs of drug crystallization, phase separation,cracking,creaming or any other signif icant changes in physical appearance after subjected to the freeze-thaw cycle and centrifugation test.All formulations which survived thermodynamic stability tests were subjected to further evaluation,including dispersibility studies, droplet size and zeta potential analysis as well as in-vitro dissolution test.

Table 2-Mean area of microemulsion region at three different ratio of surfactant to co-surfactant ratio(S/CoS).

Table 3-Selected formulations from the microemulsion region of the pseudo-ternary phase diagram constructed with lovastatin.
3.4.Precipitation and dispersibility analysis
Out of eight formulations,formulations B and C were precipitated on dilution with 0.1 N HCl at 250 times after 6 h. Although there was no sign of precipitation up to 2 h in all formulations.The capability of the self-emulsif i cation of the developed SMEDDS formulations was evaluated by dispersibility studies.Distilled water was chosen to be used as the medium to disperse the formulation since it is believed that there is no signif i cant difference if the formulations prepared with non-ionic surfactants,is dispersed in either water or simulated biological f l uids[56,57],.The results of the dispersibility test for the eight different SMEDDS formulations are summarized in Table 4.
Formulations which fell under grade C or milky appearance in dispersibility test(formulation A,B,C and D)were rejected.The remaining formulations of grade A and B(clear to bluish in appearance)namely formulation E,F,G and H were subjected to the subsequent evaluations.
3.5.Droplet size analysis
Formulation F which contains 12%of oil phase(Peceol)shown the smallest mean droplet size of<75 nm(Table 5).It was observed that as the percentage of oil phase in the formulations increases from 15%to 20%and the percentage of surfactant mixture decreases simultaneously from 85%to 80% there is a proportional increase in the mean droplet size of the formulations.This is consistent with the research studies which states that surfactants added into the microemulsion systems is mainly helps in stabilization and condensation of the interfacial f i lm which in further improves the thermodynamic stability of produced system.Presence of co-surfactant results in expansion of interfacial f i lm[58].Hence,higher ratio of surfactant to co-surfactant mixture would produce smaller droplets by decreasing the interfacial tension and consequently enabling a closely packed system[59].
Globule size is one of the most critical criteria to be evaluated in the SMEDDS formulation development.It is the key factor which affects the rate and extent of drug release[38]as well as in-vivo drug absorption in the intestine.It is reported that emulsion with smaller droplet size leads to more rapid intestinal drug absorption henceforth improves the bioavailability of the poorly absorbed drug[60].Formulation F is considered as optimized formulation.
3.6.Zeta potential analysis
The zeta potential of formulations is measured by Malvern Zetasizer.The stability of the emulsions formed is directly related to the magnitude of the zeta potential,or surfacecharge of the emulsion droplets.Zeta potential is a potential existing between the droplet surface and the dispersing liquid that will vary accordingly with the distance of the ion from the droplet surface[61].Large zeta potential of the droplets would create electrostatic repulsive forces among the droplets,thus giving the system dispersion stability to prevent coalescence of the droplets.In contrast,lower zeta potential would decrease the repulsive forces among the droplets,resulting in phase separation.It has been reported that a dividing line between stable and unstable aqueous dispersions is generally taken at either+30 or-30 mV,which means particles with zeta potentials greater than+30 mV or smaller than-30 mV could be considered stable[15,62].However,in the present study,the zeta potential of the four formulations(E,F,G and H)evaluated ranged between-10.48 to-14.89 mv as shown in Table 6.
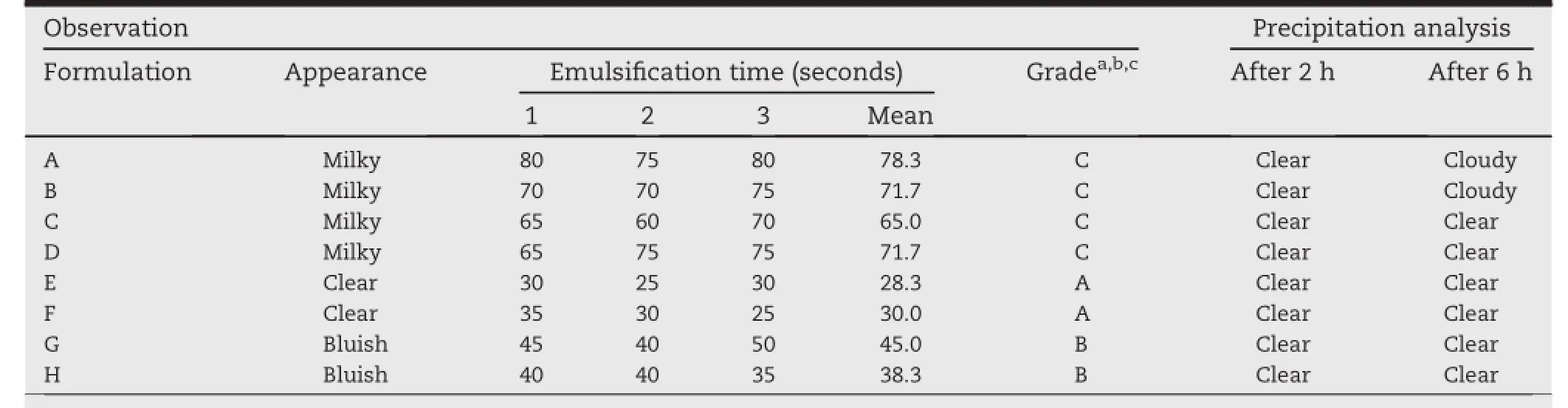
Table 4-Results of dispersibility test for selected eight different SMEDDS formulations.
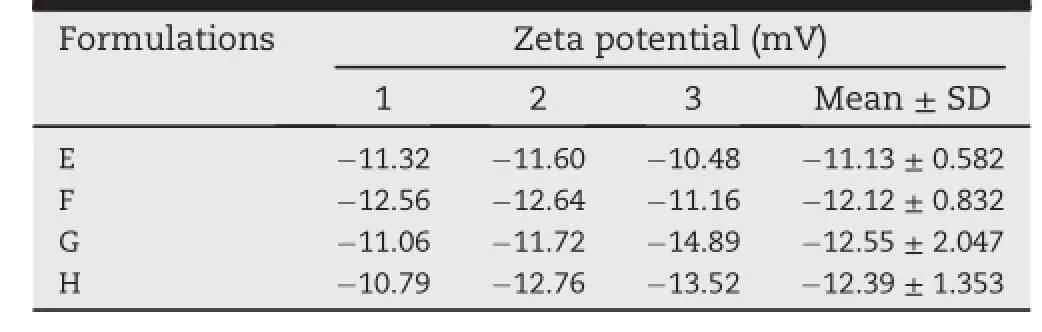
Table 6-Results of zeta potential analysis for selected four SMEDDS formulations.
3.7.In-vitro dissolution studies for developed SMEDDS
Results of release studies are presented in Fig.5 for formulation F in 0.1 NHCL and phosphate buffer pH 6.8.It can be seen clearly that lovastatin which is a lipophilic drug released at signif i cantly faster rate from SMEDDS formulations compared to pure drug.It could be suggested that SMEDDS formulation forms microemulsion with a small droplet size spontaneously upon its introduction into the dissolution media(pH 6.8 phosphatebufferand0.1 NHCl)thatpermits fasterrateofdrug release into the aqueous phase,if compared to the pure lovastatin.The mean dissolution eff i ciency(DE15min)for formulations F and pure drug was calculated to 0.9216,0.1602 in phosphate buffer pH 6.8 and 0.8143,0.0787 in 0.1 N HCl respectively.The statistical test performed on the dissolution eff i ciency(DE15min)suggests that there is statistically signif i cant difference between the dissolution rate of SMEDDS formulations,and pure lovastatin.Hence,this greater availability of dissolved form of lovastatin from the SMEDDS formulationscouldultimatelyleadtohigherintestinal absorption and thus higher oral bioavailability[38].
3.8.Preparation of LLT
Neusilin US2 is a spray dried very f i ne and ultra-light spherical granules of magnesium aluminometasilicate having high liquid absorbing capacity due to its large surface area and porous nature[35].LLT of approximately 250 mg were prepared by direct compression of Neusilin US2 with 10%crospovidone XL 10 to facilitate the disintegration and fast release of medicament.Prepared tablets have shown the porosity of 76%.Pycnometric density was determined using Accupyc 1330 Micromeritics instrument and found to be 1.87 g/cm3.Tablet density was also determined from tablet weight and volume.Tabletsporosity was found to be inversely proportional to the compression force applied.Tablets compressed with less force exhibited high porosity with suff i cient hardness required for further processing.Since tablet has to adsorb a signif i cant quantity of liquid afterward,a hardness of 6-7 kg were considered to be suitable for LLT tablets in order to have a suff i ciently hard loaded tablet f i nally.
3.9.Loading of SMEDDS in Neusilin®US2 tablets.
Amount of oil or liquid absorbed by LLT is depends on the porosity of prepared tablets,tablet with high porosity takes a large amount of oil/liquid.In this study LLT has found to absorb around 76%of lovastatin-SMEDDS.After loading the liquid,loaded tablets have shown a very low weight variation, which strongly suggests the uniform loading in tablets. However,there was slight decrease in hardness of tablet attributed to adsorption of liquid.SMEDDS loaded LLT have shown a hardness of around 4-5 kg.
3.10.Liquid loading capacity of prepared LLT incomparison of soft capsule
As per the manufacturer's specif i cation of Neusilin®US2, magnesium aluminometasilicate can absorb approximately 3 ml/g of liquid theoretically.It is reported that,after compression,tablets having porosity between 64 and 81%can absorb around 0.92-2.21 ml/g of liquid or oil[63].On the other hand,soft capsule has less loading volume because of thick shell.As per the information revealed by SGcaps®Capsugel, around 33%of volume is occupied by thick capsule shell.Hard gelatin capsule have thin wall therefore a large volume of liquid can be f i lled into these capsules but in order to avoid the risk of spillage during handling of capsules only 85-90%of total volume is f i lled with some unf i lled space.As per themanufacturer's specif i cation for hard gelatin capsule,only 72-77%volume of a capsule can be utilized for liquid f i lling (Posilok®,Qualicaps Inc.).At the same time,LLT can f i ll up to 80%of the total available volume depending on the porosity of tablet.After loading LLT can be thin coated to protect the loaded formulation.

Table 7-Effects of high cholesterol diet on rabbits lipid prof i le followed by treatment with lovastatin formulations.
3.11.In-vitro dissolution studies for developed solid SMEDDS
In-vitro dissolution test was carried out in purif i ed water as dissolution medium using USP apparatus-1(basket).Dissolution behavior of LLT was compared with capsule loaded with lovastatin suspension and capsule f i lled with lovastatin-SMEDDS.Capsule f i lled with lovastatin-SMEDDS rapidly disintegrated and released its content immediately.Lovastatin suspension has shown a very less dissolution as expected.LLT loaded with SMEDDS have shown a slower release prof i le compared to capsule f i lled with lovastatin-SMEDDS.After 1 h of dissolution approx.85%of total drug was released from LLT whereas with capsule f i lled with lovastatin-SMEDDS almost 98%drug was found to be released.Results are illustrated in Fig.6.After introducing loaded LLT into aqueous dissolution medium,loaded liquid is released into environment.Because of amphiphilic nature of SMEDDS,released liquid is easily and completely mixed with the aqueous medium.In-vitro study showed that tablets were dispersed immediately releasing formulation into dissolution media,which forms an emulsion with an average globule size of 78±5.45 nm.The globule sizeof microemulsion from LLT SMEDDS is slightly larger than the droplet size produced by liquid SMEDDS.This shows that procedure involved in making of SMEDDS-LLT does not affect the morphology of droplets of SMEDDS.Release of content from loaded LLT could be explained with convective and diffusive mechanism.Presence of crosspovidone XL 10 in tablet formula facilitates the release of drug by reducing the diffusional path length.
3.12.Shelf life determination
In order to determine the shelf life of developed optimized formulation,Accelerated stability studies were carried out at different temperature conditions(30,40 and 50°C).At the end of 60 days,concentration of drug remaining undecomposed was determined and found to be 97.12%,95.16%and 85.22%at 30°C,40°C and 50°C respectively as shown in Fig.7.Formulation were also observed for any physical change and instability and found to be stable at above stability conditions.The shelf life of the developed SMEDDS at room temperature was found to be 1.92 years.
3.13.Pharmacodynamics study
Formulation(F)optimized after in-vitro screening,was subjected for pharmacodynamics evaluation in rabbit as an animal model.The standard curve for the enzymatic determination of cholesterol,and triglyceride were prepared according to the instruction given in kit protocol.Hypercholesterolemia was induced by feeding high cholesterol diet to the rabbits.Subsequent to 4 week feeding of 2%high cholesterol diet,total cholesterol was signif i cantly increased to 1579-1723 mg/dl from 48.2 mg/dl of average normal value. SimilarlyLDLalsoelevatedto498.5-653.8g/dlfrom38.54mg/dl innormalrabbits.During4weekfeedingof2%highcholesterol diet,bloodcholesterollevelwasmaintainedelevated.Oncethe appropriate elevation in lipid prof i le was achieved,animals were divided into 6 groups as shown in Table 7,group 1 was maintained at normal diet,group 2 was maintained on high cholesterol diet whereas group 3 was maintained on placebo SMEDDS without drug but on high cholesterol diet.Remaining groups fed with high cholesterol diet were treated with lovastatinsuspension,lovastatin-SMEDDSandlovastatin-SMEDDS loaded LLT respectively.From the results obtainedof pharmacodynamics study,it was noted that lovastatin suspensionhasreducedthecholesterollevel (1578.87±94.37 mg/dl to 1427±74.77 mg/dl),triglyceride level (73.66±2.2to58.76±5.33)andLDLlevel(598.88±51.31mg/dlto 487±32.54 mg/dl)as shown in Fig.8A,B and D respectively.A slight increase in HDL level was also recorded with lovastatin suspension(Fig.8C),whereas rabbit treated with lovastatin-SMEDDS and lovastatin-SMEDDS loaded LLT have shown a signif i cantdropintheirelevatedlipidprof i le.Afteratreatment period of 4 weeks lovastatin-SMEDDS has shown to lowered down the cholesterol level from 1722.87±113.5 mg/dl to 462.44±24.7 mg/dl,LDL from 651.24±39.88 mg/dl to 175.61±33.19 mg/dl(Table 7).Similarly,lovastatin-SMEDDS loaded tablet also have shown the comparable results and a signif i cant increase in HDL level too.All the results are illustrated in Fig.8A-D.The antihyperlipidemic activity of lovastatin-SMEDDS and LLT were substantially higher(p<0.5) compared to lovastatin suspension which indicates the higher bioavailabilityorhigherplasmaconcentrationofadministered lovastatin achieved by these formulation.This improved activity of lovastatin can be explicated by the virtue of SMEDDS capability to present the drug in solubilized form which contemporarily increases the absorption of drug and thereby lead to increase plasma concentration.Poor bioavailability of lovastatin is due to its very low aqueous solubility which results in poor absorption and therapeutic activity which has beenclearlyseenwithlovastatinsuspension.Thedifferencein pharmacodynamics activity of lovastatin suspension and SMEDDS and in-vitro dissolution studies have advocated that SMEDDS formulation is capable of presenting poorly soluble druginsolubilizedformwhichmayimprovethebioavailability and therapeutic activity as well for such drug molecules. SMEDDS loaded into LLT also have shown the equivalent pharmacodynamics activity which suggests that LLT could be an appropriate and successful housing for administering SMEDDS.In this study LLT was found to absorb around 1.8 ml/ gm of liquid SMEDDS.A high loading eff i ciency is always desired in order to formulate a small solid dosage form with highliquidcontent.Attributedtohighliquidloadingcapability of these tablets,this technology is therefore recommended as better option compared to other possible substitute like, capsule f i lled SMEDDS,powder absorbed SMEDDS,liquid loaded beads etc.From commercial point of view also,this technology is easier to scale up since simple conventional method of tablet manufacturing is involved in making these liquid loaded tablets.Liquid is loaded into tablet by simple dipping method.Liquid is entrapped into tablet by capillary action which prevents the leaking of liquid[63].LLT was found to be a successful,effective and potential alternative with a high porosity and loading capability compared to other commonlyusedcarriersforSMEDDS.LLTalsohelpstoimprove thephysicalandchemicalstabilityoftheliquidformulationas it is trapped inside the solid matrix[63].
4.Conclusion
In this present study,self-emulsifying microemulsion was developed for lovastatin.Optimized SMEDDS formulation comprises of 12%peceol,44%cremophor RH 40 and 44% transcutol-P,whichshowedspontaneousemulsif i cation propertiesandgoodthermodynamicstability.SMEDDS formulation showed a better in-vitro release prof i le compared with pure drug.As per the stability studies,formulation was found to be fairly stable with expected shelf life of 1.92 years. Form pharmacodynamics studies it could be concluded that SMEDDS formulation for lovastatin has shown a signif i cantly higher lipid lowering activity compared to lovastatin suspension.At the same time,Neusilin®US2 liquid loadable tablets was also seen to be potential carrier with high loading capacity for administering liquid SMEDDS and an alternative to hard and soft gelatin capsules.
Acknowledgments
Authors are thankful to International Medical University (IMU),Malaysia for f i nancially supporting the present work under the research grant number BPharm B0108_Res322011.
REFERENCES
[1]Robinson JR.Introduction:semi-solid formulations for oral drug delivery.Bull Tech-Gattefosse 1996;89:11-13.
[2]Abdalla A,Sandra K,M¨ader K.A new self-emulsifying drug delivery system(SEDDS)for poorly soluble drugs: characterization,dissolution,in vitro digestion and incorporation into solid pellets.Eur J Pharm Sci 2008;35(5):457-464.
[3]Pouton CW.Formulation of poorly water-soluble drugs for oral administration:physicochemical and physiological issues and the lipid formulation classif i cation system.Eur J Pharm Sci 2006;29(3-4):278-287.
[4]Gursoy NR,Benita S.Self-emulsifying drug delivery systems (SEDDS)for improved oral delivery of lipophilic drugs. Biomed Pharmacother 2004;58(3):173-182.
[5]Aungst BJ.Novel formulation strategies for improving oral bioavailability of drugs with poor membrane permeation or presystemic metabolism.J Pharm Sci 1993;82:979-987.
[6]Chiou WL,Chen SJ,Athanikar N.Enhancement of dissolution rates of poorly water-soluble drugs by crystallization in aqueous surface solutions.I.Sulfathiazole,prednisone,and chloramphenicol.J Pharm Sci 1976;65:1702-1704.
[7]Pouton CW.Self-emulsifying drug delivery systems: assessment of the eff i ciency of emulsif i cation.Int J Pharm 1985a;27:335-348.
[8]Pouton CW.Effects of the inclusion of a model drug on the performance self-emulsifying formulations.J Pharm Pharmacol 1985b;37:11-17.
[9]Pouton CW.Formulation of self-emulsifying drug delivery systems.Adv Drug Deliv Rev 1997;25:47-58.
[10]Kararli TT,Needham TE,Grifaen M,et al.Oral delivery of a rennin inhibitor compound using emulsion formulation. Pharm Res 1992;9:888-893.
[11]Schwendener RA,Schott H.Lipophilic 1-beta-Darabinofuranosyl cytosine derivatives in liposomal formulations for oral and parenteral antileukemic therapy in the murine L1210 leukemia model.J Cancer Res Clin Oncol 1996;122:723-726.
[12]Srinivas C,Sagar,Vanitha S.Enhancing the bioavailability of simvastatin using microemulsion drug delivery system. Asian J Pharma Clin Res 2012;5(4):134-140.
[13]Atef E,Belmonte AA.Formulation and in vitro and in vivo characterization of a phenytoin self-emulsifying drug delivery system(SEDDS).Eur J Pharm Sci 2008;35(4):257-263.
[14]Maulik JP,Sanjay SP,Natvarlal MP.Self-microemulsifying drug delivery system.Int J Pharm Sci Rev Res 2010;4(3):29-35.
[15]Gershanik T,Benita S.Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs.Eur J Pharm Biopharm 2000;50:179-188.
[16]Kommuru T,Khan M,Reddy I.Self emulsifying drug delivery systems(SEDDS)of coenzyme Q10:formulation development and bioavailability assessment.Int J Pharm 2001;212:233-246.
[17]Jing-ling T,Jin S,Zhong-Gui H.Emulsifying drug delivery systems:strategy for improving oral delivery of poorly soluble drugs.Curr Drug Ther 2007;2:85-93.
[18]Kale AA,Vandana BP.Design and evaluation of selfemulsifying drug delivery systems(SEDDS)of nimodipine. AAPS Pharm SciTech 2008;9(1):191-196.
[19]Pouton CW.Lipid formulations for oral administration of drugs:non-emulsifying,self-emulsifying and‘selfmicroemulsifying’drug delivery systems.Eur J Pharm Sci 2000;11:93-98.
[20]Humberstone AJ,Charman WN.Lipid-based vehicles for the oral delivery of poorly water soluble drugs.Adv Drug Deliv Rev 1997;25:103-128.
[21]O'Driscoll CM.Lipid-based formulations for intestinal lymphatic delivery.Eur J Pharm Sci 2002;15:405-415.
[22]Cornaire G,Woodley J,Hermann P,et al.Impact of excipients on the absorption of P glycoprotein substrates in vitro and in vivo.Int J Pharm 2004;278(1):119-131.
[23]Jeoung HY,Shanmugam S,Thapa P,et al.Novel selfnanoemulsifying drug delivery system for enhanced solubility and dissolution of lutein.Arch Pharm Res 2010;33(3):417-426.
[24]Patel AR,Vavia PR.Preparation and in vivo evaluation of SMEDDS self-microemulsifying drug delivery system containing fenof i brate.AAPS 2007;9(3):344-352.
[25]Sha X,Yan G,Wu Y,et al.Effect of selfmicroemulsifying drug delivery systems containing labrasol on tight junctions in Caco-2 cells.Eur J Pharm Sci 2005;24:477-486.
[26]Porter CH,Trevaskis NL,Charman WN.Lipids and lipidbased formulations:optimizing the oral delivery of lipophilic drugs.Nat Rev 2007;6:231-248.
[27]Cannon JB,Long MA.Emulsions,microemulsions,and lipidbased drug delivery systems for drug solubilization and delivery-part II:oral applications.In:Liu R,editor.Waterinsoluble drug formulation.Boca Raton:CRC Press;2008. p.227-253.
[28]Bauer J,Spanton S,Henry R,et al.Ritonavir:an extraordinary example of conformational polymorphism.Pharm Res 2001;18(6):859-866.
[29]Yi T,Wan J,Xu H,et al.A new solid self-microemulsifying formulation prepared by spray-drying to improve the oral bioavailability of poorly water soluble drugs.Eur J Pharm Biopharm 2008;70(2):439-444.
[30]Patil P,Joshi P,Paradkar A.Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system(SEDDS)of ketoprofen.AAPS Pharm SciTech 2004;5(3):42-51.
[31]Dixit RP,Nagarsenker MS.Self-nanoemulsifying granules of ezetimibe:design,optimization and evaluation.Eur J Pharm Sci 2008;35(3):183-192.
[32]Nazzal S,Nutan M,Palamakula A,et al.Optimization of a self-nanoemulsif i ed tablet dosage form of ubiquinone using response surface methodology:effect of formulation ingredients.Int J Pharm 2002;240(1-2):103-114.
[33]Jannin V,Musakhanian J,Marchaud D.Approaches for the development of solid and semi-solid lipid-based formulations.Adv Drug Deliv Rev 2008;60(6):734-746.
[34]Bansal T,Mustafa G,Khan ZI,et al.Solid selfnanoemulsifying delivery systems as a platform technology for formulation of poorly soluble drugs.Crit Rev Ther Drug Carrier Syst 2008;25(1):63-116.
[35]Fuji chemical industries,technical news letter.2007.http:// www.neusilin.com/multicms/neusilin/pdf/news/29/2_ neusilin_newsletter_nov07.pdf.
[36]Lovastatin:www.rxlist.com/advicor-drug/clinicalpharmacology.htm,from 16 Dec 2013.
[37]Dhomne SG,Ajabale SB,Bhoyar GS.Formulation and evaluation of solid self emulsifying drug delivery system for lipophilic drug.Int J Pharm Sci Rev Res 2012;13(2):87-92.
[38]Bari HC,Doijad RC,More HN,et al.Design and optimization of chlordiazepoxide solid self-microemulsifying drug delivery system.J Pharm Res 2011;4(2):369-372.
[39]Yasir M,Rai S.Cinnarizine loaded lipid based system: preparation,optimization and in-vitro evaluation. J Pharmacol 2012;2(5):47-56.
[40]Lee BS,Kang MJ,Choi WS,et al.Solubilized formulation of olmesartan medoxomil for enhancing oral bioavailability. Arch Pharm Res 2009;32(11):1629-1635.
[41]Ronak PN,Tbaviskar D,Amarjit PR.Development and in-vivo characterization of SMEDDS(self-microemulsifying drug delivery system)for gemf i brozil.Int J Pharm Pharm Sci 2013;5(3):793-800.
[42]Baboota S,Abdullah,Gulam M,et al.Mechanistic approach for the development of ultraf i ne oil-water emulsions using monoglyceride and blends of medium and long chain triglycerides:enhancement of the solubility and bioavailability of perphenazine.J Excip Food Chem 2013;4(1):12-20.
[43]Balakrishnan P,Lee BJ,Oh DH,et al.Enhanced oral bioavailability of dexibuprofen by a novel solid selfemulsifying drug delivery system(SEDDS).Eur J Pharm Biopharm 2009;72(3):539-545.
[44]Bali V,Ali M,Ali J.Study of surfactant combinations and development of a novel nanoemulsion for minimizing variations in bioavailability of ezetimibe.Colloids Surf B Biointerfaces 2010;76:410-420.
[45]Shakeel F,Baboota S,Ahuja A.Stability evaluation of celecoxib nanoemulsion containing Tween 80.Thai J Pharm Sci 2008;32:4-9.
[46]Dngson P,Jangbeen K,Dajeong K,et al.Antihypercholesterolemic and anti-atherosclerotic effects of polarized light therapy in rabbits fed a high cholesterol diet. Lab Anim Res 2012;28(1):39-46.
[47]HDL and LDL/VLDL Cholesterol Assay Kit ab65390,Protocol book let,http://www.abcam.com/hdl-and-ldlvldlcholesterol-assay-kit-ab65390-protocols.html,accessed 27.01.14.
[48]Julie HC,Johnny LE,Nicole JS,et al.Molecular basis by which garlic suppresses atherosclerosis.J Nutr 2001;131:1006-1009.
[49]Adel AA,Zakaria Z,Othman F,et al.Aqueous extract of piper sarmentosum decreases atherosclerotic lesions in high cholesterolemic experimental rabbits.Lipids Health Dis 2010;9:44-52.
[50]http://www.biovision.com/manuals/K613.pdf.
[51]Groves MJ.The self-emulsifying action of mixed surfactants in oil.Acta Pharm Suec 1976;13:361-372.
[52]Shen HR,Zhong MK.Preparation and evaluation of selfmicroemulsifying drug delivery systems(SMEDDS) containing atorvastatin.J Pharm Pharmacol 2006;58:1183-1191.
[53]Liu CH,Chang FY,Hung DK.Terpene microemulsions for transdermal curcumin delivery:effects of terpenes and cosurfactants.Colloids Surf B Biointerfaces 2011;82(1):63-70.
[54]El Maghraby GM.Transdermal delivery of hydrocortisone from eucalyptus oil microemulsion:effects of cosurfactants. Int J Pharm 2008;355(1-2):285-292.
[55]Thi TD,Van SM,Barillaro V,et al.Formulate-ability of ten compounds with different physicochemical prof i les in SMEDDS.Eur J Pharm Sci 2009;38(5):479-488.
[56]Jill B,Shukla S,Patel J.Formulation and evaluation of self micro emulsifying system of candesartan cilexetil.Int J Pharm Pharm Sci 2010;2(4):143-146.
[57]Ghai D,Sinha VR.Nanoemulsions as self-emulsif i ed drug delivery carriers for enhanced permeability of the poorly water-soluble selective β1-adrenoreceptor blocker Talinolol. Nanomedicine 2012;8(5):618-626.
[58]Nazzal,Khan S,Mansoor.United States Patent 7588786, Eutectic-based self-nanoemulsif i ed drug delivery system. 2009.09/15/2009.
[59]Sierra AM,Clares B,Calpena AC,et al.Design and optimization of self-nanoemulsifying drug delivery systems (SNEDDS)for enhanced dissolution of gemf i brozil.Int J Pharm 2012;431(1-2):161-175.
[60]Shanmugam S,Baskaran R,Balakrishnan P,et al.Solid selfnanoemulsifying drug delivery system(S-SNEDDS) containing phosphatidylcholine for enhanced bioavailability of highly lipophilic bioactive carotenoid lutein.Eur J Pharm Biopharm 2011;79(2):250-257.
[61]Masoud E,Ahmad M,Elmarzugi N,et al.A novel Swietenia macrophylla oil self-nanoemulsifying system:development and evaluation.Int J Pharm Pharm Sci 2013;Supp. 5(3):639-644.
[62]Stevanovic MM,Skapin SD,Bracko I,et al.Poly(lactide-coglycolide)/silver nanoparticles:synthesis,characterization, antimicrobial activity,cytotoxicity assessment and ROS-inducing potential.Polymer 2012;53(14): 2818-2828.
[63]Sander C,Holm P.Porous magnesium aluminometasilicate tablets as carrier of a cyclosporine self-emulsifying formulation.AAPS Pharm Sci Tech 2009;10(4): 1388-1395.
*Corresponding author.Department of Pharmaceutical Technology,School of Pharmacy,Taylors University,Selangor,Malaysia.Tel.:+60 102770786.
E-mail address:mohd.javed@taylors.edu.my(M.J.Qureshi).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2014.08.003
1818-0876/©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
 Asian Journal of Pharmacentical Sciences2015年1期
Asian Journal of Pharmacentical Sciences2015年1期
- Asian Journal of Pharmacentical Sciences的其它文章
- GUIDE FOR AUTHORS
- Preparation and evaluation of taste masked oral suspension of arbidol hydrochloride
- Targeted delivery of docetaxel to the metastatic lymph nodes:A comparison study between nanoliposomes and activated carbon nanoparticles
- Degradation kinetic study of lysine in lysine hydrochloride solutions for injection by determining its main degradation product
- Preparation and evaluation of tamsulosin hydrochloride sustained-release pellets modif i ed by two-layered membrane techniques
- Evaluation of chitosan-anionic polymers based tablets for extended-release of highly watersoluble drugs