
含铋催化剂对HTPB固化反应动力学的影响
2015-05-10 01:24:09欧亚鹏常双君张百磊
含能材料 2015年6期
欧亚鹏, 常双君, 张百磊
(中北大学化工与环境学院, 山西 太原 030051)
1 引 言
端羟基聚丁二烯(HTPB)由于其生产方便、便于浇注成型,固化物具有优异的力学性能、耐低温性能等优点广泛应用于固体推进剂、浇注PBX炸药等领域[1-3]。HTPB固化反应情况直接影响其网络结构及力学性能等重要特性[4],因此Özkar[5]、王德新[6]、王晓峰[7]等人对其固化反应机理及反应动力学进行了相关研究。HTPB与固化剂的反应活性低导致装药存在固化温度高、固化时间长等缺点。采用合适的催化剂降低其固化温度,缩短固化时间是生产中切实有效的方法[8],目前常用的催化剂有二月桂酸二丁基锡(DBTDL)、乙酰丙酮铁(Fe(AA)3)、三苯基铋(TPB)等。Volkova R E[9],鲁国林[10],LUO S G[11]等针对各类催化剂的催化活性及其催化机理等做了大量研究。但这些催化剂仍存在固化温度过高的问题。实际生产中以HTPB为粘结剂的推进剂及浇注PBX炸药的固化温度约为60 ℃[12],生产过程安全性低、工艺成本高,装药冷却至室温后产生收缩应力,降低了装药力学性能。
为此,刘训恩等[13]在TPB的基础上研制了三-(乙氧基苯基)铋(TEPB),以期降低固化反应的温度,达到室温固化的目的。国内已经对TEPB催化活性进行了初步的研究[14],但大多侧重于应用研究,还未能从化学反应动力学的角度分析其催化活性。本研究采用非等温差示扫描量热法研究了TPB与TEPB催化HTPB-TDI体系固化反应的动力学,计算其活化能等动力学参数,确立其固化反应动力学方程。从动力学角度上研究了TEPB的催化活性并分析其作为室温催化剂的可能性。
2 实验部分
2.1 实验材料
端羟基聚丁二烯(HTPB),数均分子量为2800,羟值为0.78 mmol·g-1,黎明化工研究设计院有限责任公司; 2,4-甲苯二异氰酸酯(2,4-TDI),化学纯,北京化学试剂公司; 三苯基铋(TPB),化学纯,辽宁营口天元化工; 三-(乙氧基苯基)铋(TEPB),纯度(HPLC)99.54 %,中国科学院上海有机化学研究所。
2.2 实验方法
将HTPB与TDI按照固化参数为1配制,在固化体系中按实际生产中催化剂的用量[15]分别加入浓度为0.3%的TPB、TEPB两种催化剂。固化过程采用Setaram DSC131型差示扫描量热仪测量,实验条件为: N2氛围,流量为50 mL·min-1,氧化铝制样品池,温度范围为20~300 ℃,升温速率分别为5,10,15,20 ℃·min-1。样品质量约为20 mg。
3 结果与讨论
3.1 DSC测试结果
图1为升温速率为15 ℃·min-1时未加催化剂的HTPB-TDI固化体系及两种催化剂催化HTPB-TDI固化反应的DSC曲线。曲线Uncatalyzed表示未加催化剂的空白对照固化体系,曲线TPB及TEPB分别表示加入两种催化剂的固化体系。由图1可以看出HTPB的固化反应为单一的放热反应,加入催化剂降低了反应温度,使DSC曲线中的放热峰向低温方向移动。

图1 升温速率为15 ℃·min-1时各固化体系的DSC曲线
Fig.1 DSC curves of different curing system at 15 ℃·min-1
加入TPB的固化体系中,除有一个明显的放热峰外,后期还有一个很微弱的放热峰,并且只有在升温速率为15 ℃·min-1的时候才能观察到。而在其他升温速率时,DSC曲线上未表现出后期的放热峰,很可能是后期的反应放热被第一个放热峰覆盖了。TPB反应机理[11]表明,TPB中的铋原子与羟基氢首先络合,强化了羟基氧的亲核性,TPB与羟基氢间的作用会使部分氢键缔合态的羟基转化为自由态,而自由态羟基的氢更易与TPB中碱性的铋原子形成络合作用,使羟基氧的亲核性增强,更容易进攻异氰酸根中的碳原子,形成过渡态; 或者直接活化体系中存在的羟基-异氰酸根缔合物,碱性的铋原子与羟基氢络合强化了羟基氧的亲核性,使反应更易进行,最终生成产物氨基甲酸酯。TPB在形成氨基甲酸酯后会从与羟基形成的络合物中脱出,而后一个放热峰之所以不明显,甚至被第一个放热峰覆盖说明TPB从络合物中脱出的反应放热很小,并且反应几乎是与凝胶反应同时进行的,因此无法单独计算其反应动力学,所以在此只讨论其前期形成凝胶的化学反应动力学。
表1和表2是HTPB-TDI和HTPB-TDI-TPB固化体系DSC曲线对应的特征峰温。加入TPB的固化体系峰顶温度Tp较未加催化剂平均降低了75 ℃,并且随着升温速率的增大,峰顶温度差也逐渐增加; 未加催化剂的固化体系中固化起止温差(Tf-Ti)约为50 ℃,而TPB的加入使(Tf-Ti)降低至约20 ℃,催化剂降低了固化反应的温度差,减少了反应时间。说明TPB可有效地降低HTPB的固化温度并缩短固化时间。
表1 HTPB-TDI固化反应的DSC曲线特征峰温
Table 1 DSC characteristic temperatures of HTPB-TDI curing reaction
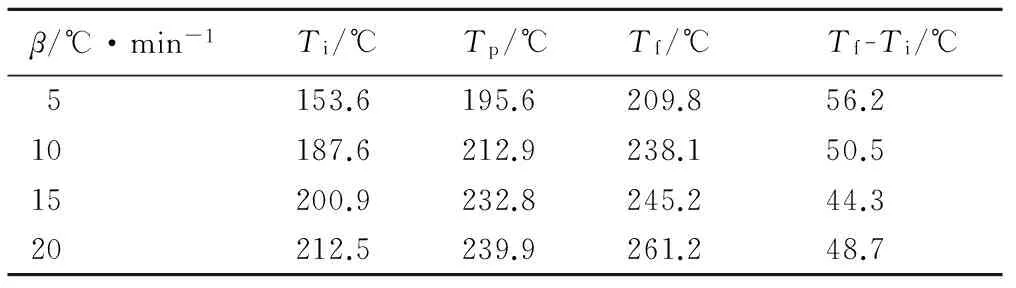
β/℃·min-1Ti/℃Tp/℃Tf/℃Tf-Ti/℃5153.6195.6209.856.210187.6212.9238.150.515200.9232.8245.244.320212.5239.9261.248.7
Note:β, heating rate;Ti, the initial curing temperature;Tp, the peak curing temperature;Tf, the end curing temperature.
表2 TPB催化HTPB-TDI固化反应的DSC曲线特征峰温
Table 2 DSC characteristic temperatures of TPB catalysed HTPB-TDI reaction
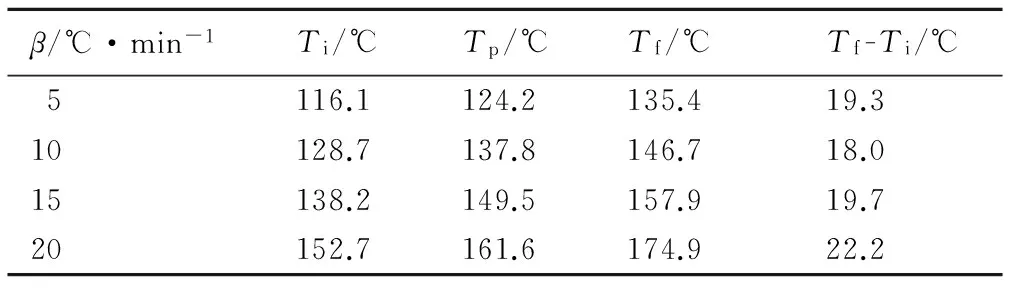
β/℃·min-1Ti/℃Tp/℃Tf/℃Tf-Ti/℃5116.1124.2135.419.310128.7137.8146.718.015138.2149.5157.919.720152.7161.6174.922.2
与TPB不同的是,加入TEPB的固化体系表现出两个明显的放热峰,其原因为第一个放热峰的形成是由于TEPB首先与羟基络合,并与异氰酸酯基形成过渡态,即HTPB与TDI发生凝胶反应的过程; 第二个放热峰是由于TEPB与过渡态的羟基-异氰酸根缔合物不稳定,催化剂从与羟基形成的络合物中脱出所造成的。TEPB是在TPB的基础上引入了乙氧基,由于乙氧基电子云密度大,是给电子基,因此与羟基络合后较TPB更大地增加了其电负性,使其更容易与异氰酸酯基发生反应,这就是TEPB催化活性高于TPB的原因。
TEPB催化HTPB-TDI固化从DSC曲线上可以分为明显的前后两个阶段。TEPB催化HTPB-TDI固化前期及后期反应的DSC曲线特征峰温见表3。反应前期的固化体系峰顶温度较未加催化剂平均降低了90 ℃; TEPB使固化反应前期的(Tf-Ti)约为30 ℃,高于TPB催化体系的20 ℃。说明TEPB能较大地降低固化反应的温度,但缩短反应时间方面较TPB差。而后期反应的温度高说明TEPB与羟基-异氰酸根形成的过渡态较稳定。不同催化剂的催化机理虽然存在显著差异,但过渡态越稳定表现为该体系的反应速率越大,催化剂的催化活性越高。而TPB后期反应的温度低,甚至被第一个放热峰覆盖说明其生成的过渡态不稳定,这也从反应机理的角度说明了TEPB的催化活性高于TPB。
表3 TEPB催化HTPB-TDI固化前期及后期反应的DSC曲线特征峰温
Table 3 DSC characteristic temperatures of early and later stage of TEPB catalysed HTPB-TDI reaction
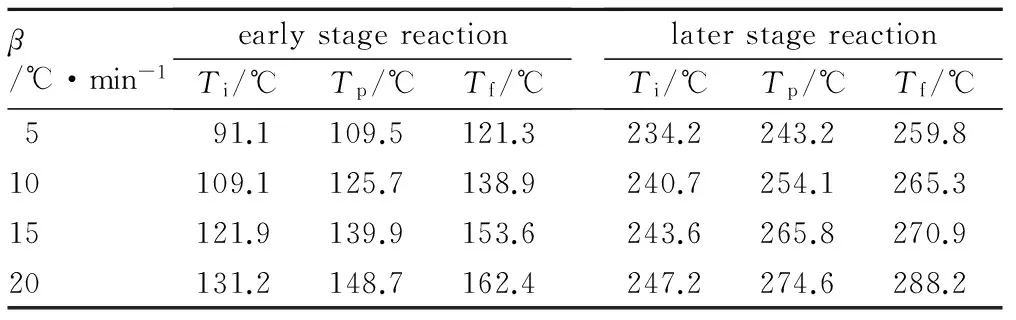
β/℃·min-1earlystagereactionTi/℃Tp/℃Tf/℃laterstagereactionTi/℃Tp/℃Tf/℃591.1109.5121.3234.2243.2259.810109.1125.7138.9240.7254.1265.315121.9139.9153.6243.6265.8270.920131.2148.7162.4247.2274.6288.2
3.2 催化剂催化HTPB-TDI体系的固化反应动力学
计算固化反应体系的动力学参数采用有Kissinger[16]法和Crane[17]法。
Kissinger公式:
(1)
Crane公式:
d(lnβ) /d(Tp-1)=-(Ea/nR+2Tp)
(2)
Ea/nR≫2Tp, d( lnβ)/d(Tp-1) =-Ea/nR
式中,Ea是固化反应活化能,kJ·mol-1;R是气体常数,8.314 J·K-1·mol-1;T是绝对温度,K;n为反应级数。
对HTPB固化反应机理的研究表明HTPB-TDI体系的固化反应遵循n级反应模型[18-19],其反应动力学模型方程如下:
dα/dt=k(1-α)n
(3)
式中,α是固化度;t是时间,s; dα/dt是固化速率,s-1;k是反应速率常数,s-1,服从Arrhenius关系
k=Aexp(-Ea/RT)
(4)


表4 各固化体系的动力学参数
Table 4 Kinetic parameters of different curing systems
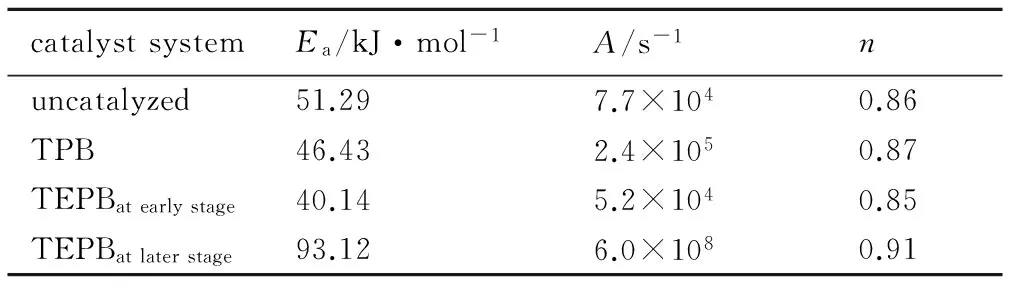
catalystsystemEa/kJ·mol-1A/s-1nuncatalyzed51.297.7×1040.86TPB46.432.4×1050.87TEPBatearlystage40.145.2×1040.85TEPBatlaterstage93.126.0×1080.91
将上述参数代入方程(3)和(4)可得:
HTPB-TDI体系的固化反应动力学方程为:
dα/dt=7.7×104exp(-6169/T)( -α)0.86;
(5)
HTPB-TDI-TPB体系的固化反应动力学方程为:
dα/dt=2.4×105exp(-5584/T)(1-α)0.87;
(6)
HTPB-TDI-TEPB体系的固化反应前期动力学方程为:
dα/dt=5.2×104exp(-4828/T)(1-α)0.85;
(7)
HTPB-TDI-TEPB体系的固化反应后期动力学方程为:
dα/dt=6.0×108exp(-11199/T)(1-α)0.91。
(8)
TPB催化体系的A大于未加催化剂的固化体系,说明TPB不仅降低了固化反应的活化能,且增大了分子的碰撞频率从而加快了反应速率,这是TPB能有效降低反应温度,缩短固化时间的内在原因。TEPB进一步降低了反应的活化能,但A小于TPB催化体系,这也是其能较大程度地降低反应温度,固化时间却较TPB催化体系长的原因。表4结果说明三个固化体系的反应级数基本一致,说明催化剂并不能改变其基本的基元反应,固化反应能较好地符合n级反应模型。
由以上动力学方程可知,固化反应速率常数k与反应温度T呈线性关系,反应速率常数决定了该温度下固化反应速率的快慢。表5是部分温度下各催化体系的反应速率常数。实际生产中以TPB为催化剂的HTPB-TDI粘结剂的固化温度为50~60 ℃。当固化温度为60 ℃时,加入TPB作催化剂的固化体系反应速率常数为1.25×10-2s-1,高于未加催化剂时121 ℃的k值,但聚氨酯固化温度一般都在100 ℃以下,温度过高会导致异氰酸酯基团与氨基甲酸酯或脲键反应,产生交联键,降低聚氨酯的性能; 而且高温会严重影响推进剂与浇注PBX炸药生产过程中的安全性。这也是在固化过程中加入催化剂降低其固化温度的重要原因。而以TEPB作催化剂的固化体系在44 ℃时反应速率常数为1.26×10-2s-1,即达到了加入TPB催化时60 ℃的k值,这说明TEPB的加入可以降低了反应温度。加入TEPB的固化体系在34 ℃时的反应速率常数即可高于使用TPB作为催化剂时50 ℃时的反应速率常数。此数据从反应动力学角度证明了TEPB作为室温(25~35 ℃)固化催化剂的可行性。
表5 不同温度下各催化体系中的反应速率常数
Table 5 Reaction constants of different catalyst system under different temperature
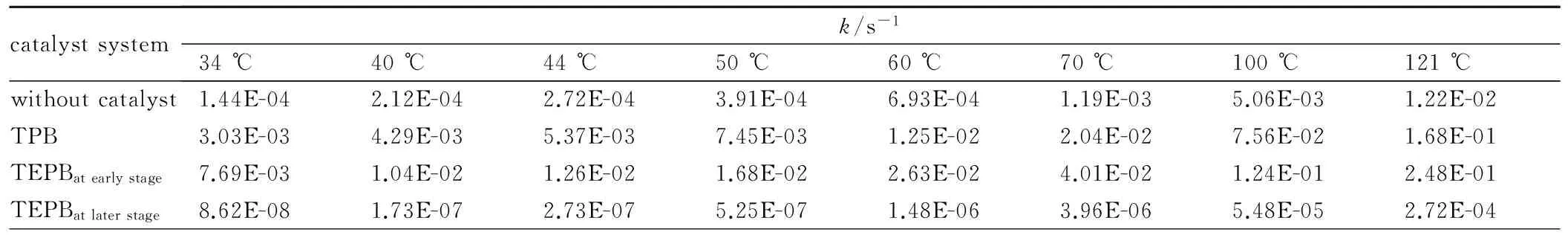
catalystsystemk/s-134℃40℃44℃50℃60℃70℃100℃121℃withoutcatalyst1.44E-042.12E-042.72E-043.91E-046.93E-041.19E-035.06E-031.22E-02TPB3.03E-034.29E-035.37E-037.45E-031.25E-022.04E-027.56E-021.68E-01TEPBatearlystage7.69E-031.04E-021.26E-021.68E-022.63E-024.01E-021.24E-012.48E-01TEPBatlaterstage8.62E-081.73E-072.73E-075.25E-071.48E-063.96E-065.48E-052.72E-04
4 结 论
(1)无催化剂的HTPB-TDI固化体系的Ea=51.29 kJ·mol-1,HTPB-TDI-TPB固化体系的Ea=46.43 kJ·mol-1,HTPB-TDI-TPB固化体系反应前期的Ea=40.14 kJ·mol-1,催化剂的加入降低了反应的活化能,降低了反应温度。
(2)HTPB-TDI-TPB体系的固化反应动力学方程为: dα/dt=2.4×105exp(-5584/T)(1-α)0.87,HTPB-TDI-TEPB体系的固化反应前期动力学方程为: dα/dt=5.2×104exp(-4828/T)(1-α)0.85,TEPB催化HTPB固化反应速率大于TPB催化反应速率,TEPB催化活性较高。
(3)加入TEPB的固化体系在34 ℃时反应速率常数即高于加入TPB的固化体系在50 ℃时的反应速率常数。TEPB可在室温条件下催化HTPB-TDI固化。
参考文献:
[1] WANG Jing-yu, AN Chong-wei, LI Gang,et al. Preparation and performances of castable HTPB/CL-20 booster explosives[J].Propellants,Explosives,Pyrotechnics, 2011, 36: 34-41.
[2] Kumar S Adapaka, Rao B Vepakomma, Sinha K Rabindra,et al. Evaluation of plastic bonded explosive (PBX) formulations based on RDX, aluminum, and HTPB for underwater applications[J].Propellants,Explosives,Pyrotechnics, 2010, 35: 359-364.
[3] Nanda KumarJogesh and Ramakrishna A P. Development of AP/HTPB based fuel-rich propellant for solid propellant ramjet[C]∥AIAA paper 2013: 4149-4171.
[4] Ahmad Nadeem, M B Khan, Ma XY,Ul-Haq Noaman and Ihtasham-ur-Rehman. Dynamic mechanical characterization of the crosslinked and chainextended HTPB based polyurethanes[J].Polymers&PolymerComposites, 2012,20: 683-691.
[5] DKincal, S Özkar. Kinetic study of the reaction between hydroxyl-terminated polybutadiene and isophorone diisocyanate in bulk by quantitative FTIR spectroscopy[J].JournalofAppliedPolymerScience,1997,66(10): 1979-1983.
[6] 王新德, 张捷, 刘艳艳, 等. IPDI与HTPB反应动力学傅里叶红外研究[J]. 化学推进剂与高分子材料,2011,9(6): 64-68.
WANG Xin-de, ZHANG Jie, LIU Yan-yan,et al.Researches on IPDI and HTPB reaction kinetics by foruier transform infrared spectrum[J].ChemicalPropellants&PolymericMaterials(HannengCailiao),2011,9(6): 64-68.
[7] 陈春燕, 王晓峰, 高立龙, 等. 不同分子量HTPB与TDI的固化反应动力学[J]. 含能材料, 2013, 21(6): 771-776.
CHEN Chun-yan, WANG Xiao-feng, GAO Li-long, et al. Effect of HTPB with different molecular weights on curing kinetics of HTPB/TDI system[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2013, 21(6): 771-776.
[8] Korah C Bina, Kannan G K, Ninan.N K DSC study on the effect of isocyanates and catalysts on the HTPB cure reaction[J].JournalofThermalAnalysisandCalorimetry, 2004, 78(3): 753-760.
[9] Volkova R E,Tereshatov V V,and Karmanov I V. Influence of the concentration of iron(III) tris(acetylacetonate) used as urethane formation catalyst on the structure and properties of cold-cure polyurethanes[J].RussianJournalofAppliedChemistry,2011, 84(8): 1414-1417.
[10] 鲁国林, 夏强, 杜娟. 三苯基铋对高燃速丁羟推进剂的催化固化作用研究[J]. 含能材料,1999, 7(2): 60-61.
LU Guo-lin,XIA Qiang,DU Juan.Catalysis effect of TPB on the cure of HTPB-based high burning rate propellant[J].ChineseJournalofEnergeticMaterials(HannengCailiao),1999,7(2): 60-61.
[11] LUO S G,TAN H M,ZHANG JG,et al. Catalytic mechanisms of triphenyl bismuth,dibutyltin dilaurate,and their combination in polyurethane-forming reaction[J].JournalofAppliedPolymerScience,1997,65(6):1217-1225.
[12] Thibieroz B, Lecume S, Bigot Y. Development and characterization of PBX cast at ambient temperature. Insensitive Munitions and Energetic Materials Technology Symposium[C]∥2001: 531-542.
[13] 刘训恩, 缪琳, 陈力, 等. 三-(乙氧基苯基)铋的合成及应用[J]. 北京理工大学学报, 1995, 15(6): 7-9.
LIU Xun-en, MIAO Lin, CHEN Li, et al. Synthesis and application of tris-(ethoxyphenyl) bismuthine[J].JournalofBeijingInstituteofTechnology, 1995, 15(6): 7-9.
[14] 刘训恩, 唐松青. 室温固化催化剂的研制和在固体推进剂中的应用[J]. 化学推进剂与高分子材料, 2004, 2(2): 4-6.
LIU Xun-en, TANG Song-qing. Development of ambient temperature curing catalysts and their applications in solid propellants[J].ChemicalPropellants&PolymericMaterials, 2004, 2(2): 4-6.
[15] 鲁国林, 夏强, 杜娟. 丁羟推进剂粘合剂体系固化催化研究[J]. 推进技术,1998,19(6): 97-100.
LU Guo-lin, XIA Qiang, DU Juan. Study on curing catalysis of HTPB propellant binder system[J].JournalofPropulsiontechnology, 1998, 19(6): 97-100.
[16] JaroslavSestak. Is the original Kissinger equation obsolete today: not obsolete the entire non-isothermal kinetics[J].JournalofThermalAnalysisandCalorimetry,2014, 117(1): 3-7.
[17] Crane W L, Dynes J P,DKaelble H. Analysis of curing kinetics in polymer composites[J].PolymerLetterEdition,1973,11: 533.
[18] Singh M,Kanungo K B, Bansal K T. Kintic studies on curing of hydroxy-terminated polybutadiene prepolymer-based polyurethane networks[J].JournalofAppliedPolymerScience, 2002, 85(4): 842-846.
[19] Han J L, Yu C H, Lin Y H, et al. Kinetic study of the urethane and urea reactions of isophorone diisocyanate[J].JournalofAppliedPolymerScience,2008,107(6): 3891-3902.
[20] 陈清元, 陈中华, 程时远, 等. HTPB/TDI的反应动力学研究[J]. 高分子材料科学与工程,1996,12(3):45-49.
CHEN Qing-yuan, CHEN Zhong-hua, CHENG Shi-yuan, et al. Study on reaction kinetics of HTPB/TDI by FT-IR[J].PolymericMaterialsScienceandEngineering,1996,12(3):45-49.
猜你喜欢
数学年刊A辑(中文版)(2021年1期)2021-06-09 09:32:06
中成药(2018年2期)2018-05-09 07:20:05
新乡学院学报(2016年6期)2016-12-01 05:21:38
山西大同大学学报(自然科学版)(2016年4期)2016-11-27 02:20:55
新高考·高一物理(2016年3期)2016-05-18 16:16:56
当代化工研究(2016年9期)2016-03-20 16:22:11
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
无机化学学报(2014年4期)2014-02-28 17:31:23
应用技术学报(2014年1期)2014-02-28 14:52:11
