
Synthesis and Crystal Structure of 6b-Nitrohexahydro-2H-1,3,5-trioxacyclopenta[cd]-pentalene-2,4,6-triyl trinitrate
2015-05-10 00:33:23LIXiangzhiLIYananZHOUChengLIHuiFANXuezhongWANGBozhou
含能材料 2015年4期
LI Xiang-zhi, LI Ya-nan, ZHOU Cheng, LI Hui, FAN Xue-zhong, WANG Bo-zhou
(Xi′an Modern Chemistry Research Institute, Xi′an 710065, China)
1 Introduction
2 Experimental
2. 1 Reagents and measurements
All chemical reagents from commercial sources were of analytically pure grade except for the industrial conc. sulfuric acid(95%-98%) and nitric acid(98%).Melting point was measured on a X-6 Melting-Point Apparatus with Microscope. Elemental analysis was performed by a Vario EL Ⅲ elementary analysis instrument. IR spectra were recorded on a Nexus 870 FT-IR instrument using KBr pellet in the 4000-400 cm-1. NMR spectrum was obtained on a Bruker AV 500-MHz spectrometer with tetramethylsilane(TMS) as the internal standard and acetone-d6as the solvent.
2.2 Synthesis of the title compound

Scheme 1
2.2.1 Synthesis of 6b-Nitrohexahydro-2H-1,3,5-trioxacyclopenta[cd]-pen- talene-2,4,6-triol(1)
Nitromethane(2.5 mL, 47 mmol) was added to aqueous glyoxal(40%, 8 mL, 70 mmol), and then aqueous solution of sodium hydroxide(10%, 2 mL) was added dropwise at 15-20 ℃. The mixture was kept at room temperature for 2 h and the precipitated product was filtered off, washed with water. After crystallization from water and drying, the 6b-nitrohexahydro-2H-1,3,5-trioxacyclopenta[cd]-pen- talene-2,4,6-triol(1) was obtained with yield of 60%. m.p.: 152-153 ℃,1H NMR (DMSO-d6,500 MHz),δ: 4.94(s, 3H, H2a, H4a, H6a), 5.37(d,J=3.5Hz, 3H, H2, H4, H6), 6.96(d,J=3.75, 3H, OH);13C NMR (DMSO-d6,125 MHz) 91.16(C2a, C4a, C6a), 102.45(C2, C4, C6), 106.85(C6b); IR(KBr,ν/cm-1): 3318(—OH), 2978(—CH), 1556, 1356(—NO2); Anal. Calcd for C7H9NO8: C 35.75, H 3.86, N 5.96 ; Found: C 34.90, H 3.73, N 5.50.
2.2.2 Synthesis 6b-nitrohexahydro-2H-1,3,5-trioxacyclopenta[cd]-pentalene-2,4,6-triyl trinitrate(2)
Nitric acid(2 mL, 45 mmol) was added to conc. sulfuric acid(4.8 mL, 90 mmol), and then compound1(2.34 g, 10 mmol) was added slowly at 0-5℃, with vigorously stirring. The mixture was kept at this temperature for 45 min and poured over crushed ice. A solid precipitate was filtered off and washed with water until to neutral. After the crystallization from methanol, pure compound 2 was obtained with the yield of 68.16%, m.p. 119-120 ℃.1H NMR (acetone-d6, 500 MHz),δ: 5.64(s, 3H, H2a, H4a, H6a), 6.61(s, 3H, H2, H4, H6);13C NMR (acetone-d6, 125 MHz),δ: 91.08(C2a, C4a, C6a), 104.90(C6b), 106.9(C2, C4, C6),14N NMR (acetone-d6, 36.13 MHz ),δ: 284.11 (ν1/2=1450 Hz), 325.15(ν1/2=750 Hz),15N NMR(acetone-d6,50.67 MHz ),δ: 327.76, 368.4; IR(KBr,ν/cm-1): 2978(—CH),1571, 1306(—NO2); Anal. Calcd for C7H6N4O14: C 22.71, H 1.63,N 15.14 ; Found: C 23.00, H 1.61, N 14.95.
2.3 X-ray crystal structure determination
The title compound 2 was dispersed into ethanol/n-butyl alcohol (V/V=1/1) with a stirrer, and the impurities were eliminated by filtration. The saturated solution of compound 2 was put in a conical flask for two weeks at room temperature staticly, and evaporated slowly to obtain single crystal of compound 2.
A colorless single crystal of compound 2 with dimensions of 0.37 mm×0.31 mm×0.25 mm was selected for X-ray single diffraction analysis, and the data collection was performed on a Bruker SMART Apex II CCD X-ray diffractometer equipped with a graphite-monochromatized Mo Kαradiation (λ=0.71073 Å) using theφ-ωscan mode (2.12°<θ<25.10°) at 296(2) K. A total of 6234 reflections were collected, of which 2326 were independent (Rint=0.0426) and 1986 withI>2σ(I) were considered to observed and used for the refinement.
The structure was solved by direct methods and refined by full-matrix least squares techniques onF2using SHELXS-97 and SHELXL-97 programs[8-10]. All non-hydrogen atoms were refined anisotropically. The atomic scattering factors and anomalous dispersion corrections were taken from X-ray crystallography[8]. The final refinement gaveR=0.488,wR=0.1289(w=1/[σ2(Fo2)+(0.0708P)2+0.4691P, whereP=(Fo2+2Fc2)/3),S=1.097, (/ó)max=0.001, ()max=0.478 and ()min=-0.266 e/Å3. Crystal data and structural refinement parameters for compound 2 were listed in Table 1.
3 Results and discussion
3.1 Synthesis of title compound
Using nitromethane and glyoxal as the starting materials, compound 2 was synthesized by the process of condensation and nitration reactions, and the total yield could reach 40.90%. Stanistaw[7]synthesized compound 2 using nitric acid as the nitration reagent with 55% yield. In order to increase the yield of nitration reaction, the different nitration agents were used and the results were given in Table 2. The product wasn′t found with a mixture of potassium nitrate in conc. sulfuric acid as the nitration reagent. The mixture of nitric acid and conc. sulfuric acid were strong nitration agent and gave the target product in a yield of 68.16%. The elemental analysis, IR,1H NMR,13C NMR,14N NMR and15N NMR for the product are in good agreement with the title compound.
Table 1 Crystaldata of compound 2

formulaC7H6N4O14formulamass370.16crystalsystemMonoclinicspacegroupP2(1)/na/Å7.792(9)b/Å8.534(9)c/Å19.41(2)β/(°)117.624(4)crystalsize/mm30.37×0.31×0.25volume/Å31305(2)Z4Dc/g·cm-31.884F(000)752T/K296(2)λ/Å0.710732θ/(°)50.20reflectionscollected/unique6234/2326parameters227GOFonF21.097finalR1,wR2(I>2σ(I))0.0488,0.1289finalR1,wR2(alldata)0.0567,0.1348largestdiffpeakandhole/e·Å-30.478and-0.266
Table 2 Influence of the nitration reagent on the yield of compound 2
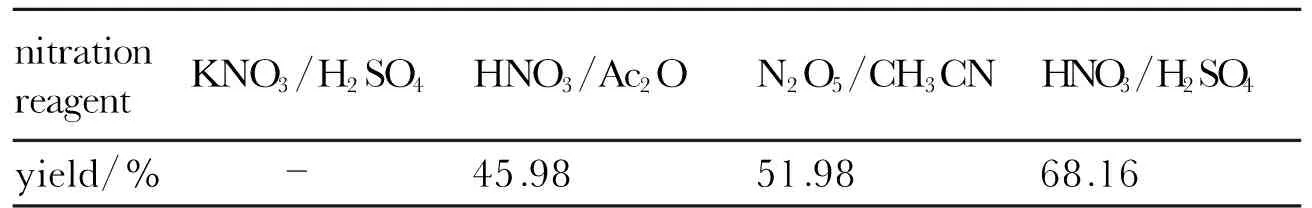
nitrationreagentKNO3/H2SO4HNO3/Ac2ON2O5/CH3CNHNO3/H2SO4yield/% -45.9851.9868.16
3. 2 Crystal structure
The molecular structure and packing diagram of compound 2 are depicted in Fig.1 and Fig.2. The the bond lengths and angles of the compound is presented in Table 3 and Table 4, which shows in the molecule are within normal ranges[11].The structure of compound 2 is rotational symmetry, which looks like a red-wine glass with a nitro on the bottom and three nitric acid esters in flank. There are three tetrahydrofuran rings in the molecular. Moreover, the four carbon atoms of the tetrahydrofuran ring are nearly in a plane (the dihedral angles of C(1)—C(6)—C(7)—C(2) is -10.2°). The dihedral angles between plane defined by C(1)—O(12)—C(2) and plane C(2)—C(7)—C(6), between plane by C(1)—C(6)—C(7) and plane C(2)—C(7)—C(6) are 36.16° and 26.16°, respectively.
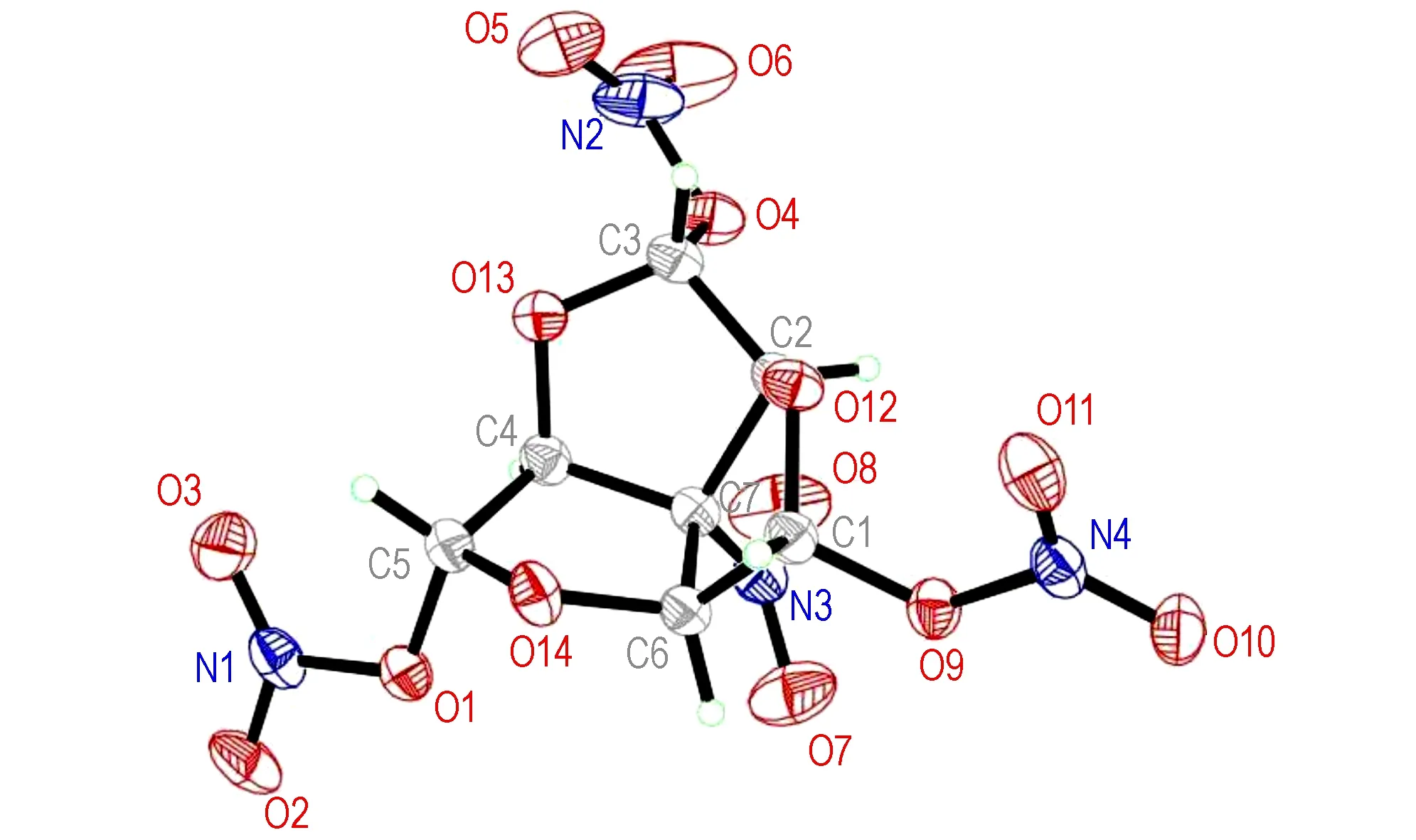
Fig.1 Crystal structure of compound 2
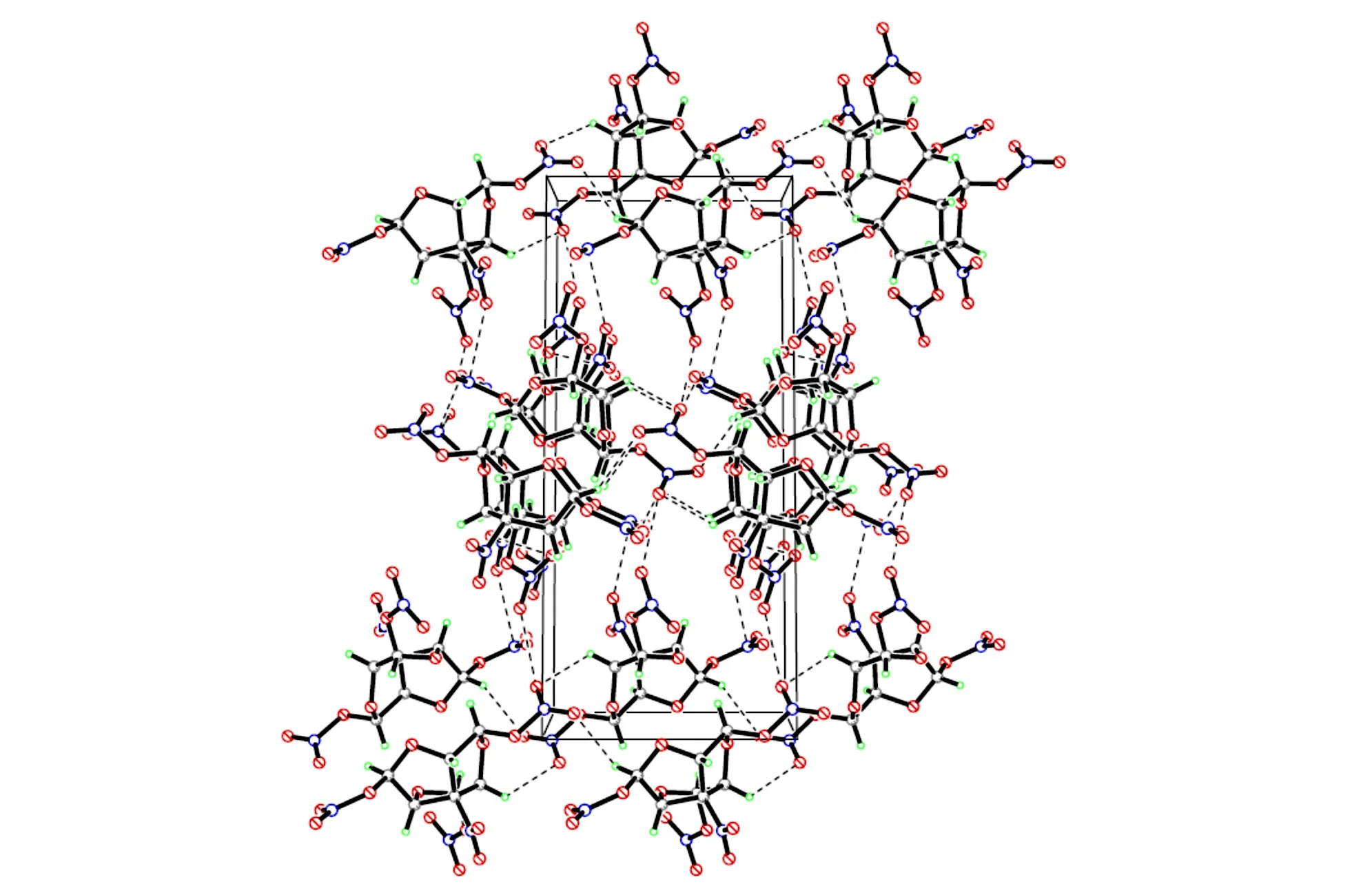
Fig.2 Molecular packing diagram of the unit cell of compound 2
Table 3 Selected bond lengths of compound 2
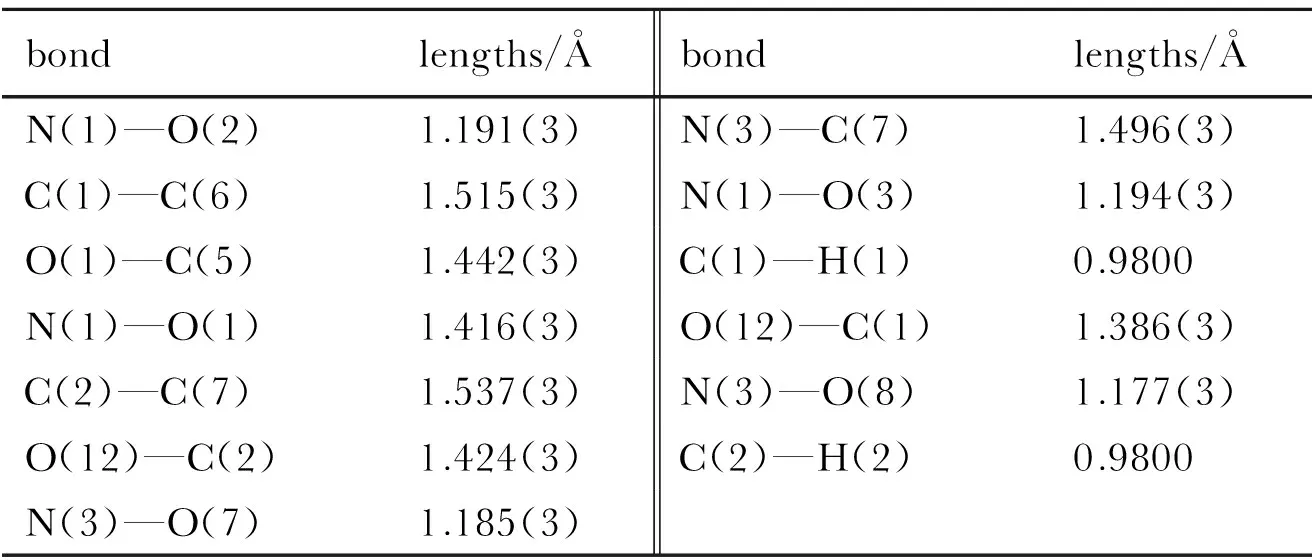
bondlengths/Åbondlengths/ÅN(1)—O(2)1.191(3)N(3)—C(7)1.496(3)C(1)—C(6)1.515(3)N(1)—O(3)1.194(3)O(1)—C(5)1.442(3)C(1)—H(1)0.9800N(1)—O(1)1.416(3)O(12)—C(1)1.386(3)C(2)—C(7)1.537(3)N(3)—O(8)1.177(3)O(12)—C(2)1.424(3)C(2)—H(2)0.9800N(3)—O(7)1.185(3)
Table 4 Selected bond angles and torsion angles of compound 2

bondangle/(°)O(2)—N(1)—O(3)131.5(3)O(12)—C(1)—C(6)106.35(18)C(3)—C(2)—H(2)113.4O(2)—N(1)—O(1)111.5(2)O(9)—C(1)—C(6)103.96(18)C(7)—C(2)—H(2)113.4O(3)—N(1)—O(1)117.0(2)O(12)—C(1)—H(1)111.5N(3)—C(7)—C(6)114.3(2)O(8)—N(3)—O(7)122.5(2)O(9)—C(1)—H(1)111.5N(3)—C(7)—C(4)114.20(18)O(8)—N(3)—C(7)117.3(2)C(6)—C(1)—H(1)111.5C(6)—C(7)—C(4)105.24(17)O(7)—N(3)—C(7)120.1(2)O(12)—C(2)—C(3)106.47(18)

bondangle/(°)N(3)—C(7)—C(2)112.28(18)N(1)—O(1)—C(5)113.74(18)O(12)—C(2)—C(7)105.09(16)C(6)—C(7)—C(2)105.40(17)C(1)—O(12)—C(2)109.29(16)C(3)—C(2)—C(7)104.29(19)C(4)—C(7)—C(2)104.47(19)O(12)—C(1)—O(9)111.67(18)O(12)—C(2)—H(2)113.4O(2)—N(1)—O(1)—C(5)-176.9(2)C(7)—C(2)—C(3)—O(13) 25.0(2)C(1)—C(6)—C(7)—N(3)113.6(2)O(3)—N(1)—O(1)—C(5) 3.9(3)O(12)—C(2)—C(3)—O(4)154.87(17)C(1)—C(6)—C(7)—C(4)-120.28(19)C(2)—O(12)—C(1)—O(9) 78.3(2)C(7)—C(2)—C(3)—O(4)-94.3(2)O(14)—C(6)—C(7)—C(2)102.0(2)C(2)—O(12)—C(1)—C(6)-34.5(2)O(12)—C(1)—C(6)—O(14)-84.4(2)C(1)—C(6)—C(7)—C(2)-10.2(2)N(4)—O(9)—C(1)—O(12) 72.5(2)O(9)—C(1)—C(6)—O(14)157.58(16)O(13)—C(4)—C(7)—N(3)-130.52(19)N(4)—O(9)—C(1)—C(6)-173.25(16)O(12)—C(1)—C(6)—C(7) 26.7(2)O(13)—C(4)—C(7)—C(2) -7.5(2)C(1)—O(12)—C(2)—C(3)137.40(18)O(9)—C(1)—C(6)—C(7)-91.3(2)O(12)—C(2)—C(7)—C(6) -8.9(2)C(1)—O(12)—C(2)—C(7) 27.1(2)O(8)—N(3)—C(7)—C(6)-172.2(3)O(12)—C(2)—C(7)—C(4)101.72(19)O(12)—C(2)—C(3)—O(13)-85.8(2)O(7)—N(3)—C(7)—C(6) 4.1(3)
Table 5 Hydrogen bond lengths and bond angles
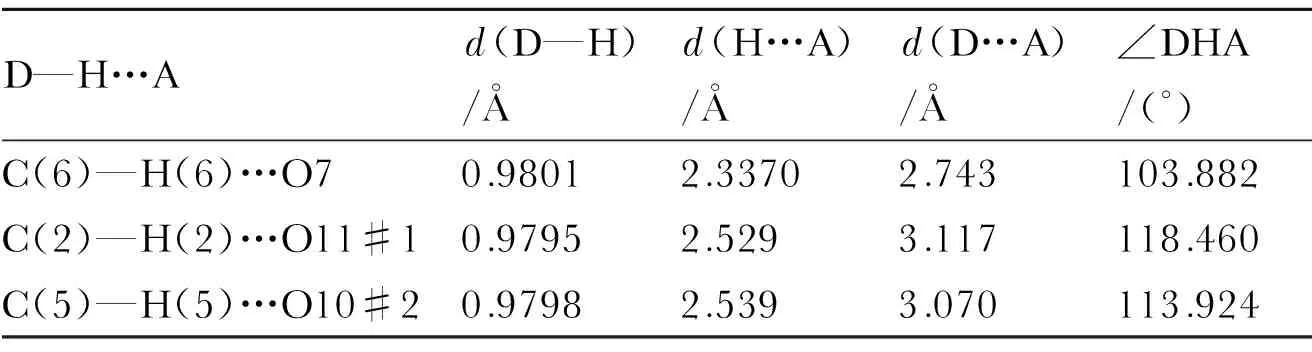
D—H…Ad(D—H)/Åd(H…A)/Åd(D…A)/Å∠DHA/(°)C(6)—H(6)…O70.98012.33702.743103.882C(2)—H(2)…O11#10.97952.5293.117118.460C(5)—H(5)…O10#20.97982.5393.070113.924
Note: Symmetry codes, #1 -x+1, -y+2, -z+2; #2x, -y+1,z.
4 Conclusions
The title compound was synthesized by using the mixture of nitric acid and conc. sulfuric acid instead of merely nitric acid. It can be indicated from X-ray single-crystal diffraction analysis that the structure of compound 2 is rotational symmetry with three tetrahydrofuran rings, three nitric acid esters and a nitro. Due to the weak intermolecular contacts, intramolecular and intermolecular hydrogen bonds, the density of compound 2 could reach up to 1.884 g·cm-3.
[1] Jeong K K, Jin S K, Keun D L. A new energetic mixed formal plasticizer, using diformal as energetic material[C]∥Insensitive Munitions and Energetic Materials Technology Symposium. SanDiego, 2000: 421-426.
[2] LUO Yun-jun, LIU Jing-ru. Researsh progress of high energy solid propellant[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2007, 15(4): 407-410.
[3] LIU Jing-ru, LUO Yun-jin, YANG Yan. Energetic characteristics calculaton of a new generation high energetic solid propellant[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2008, 16(1): 94-99.
[4] JI Yue-ping, LI Pu-rui, WANG Wei,et al. A review of recent advances of energetic plasticizers[J].ChineseJournalofExplosives&Propellants, 2005, 28(4): 47-51.
[5] BI Fu-qiang, WANG Bo-zhou, FAN Xue-zhong, et al. Synthesis and structural characterization of 2,3-bis(hydroxymethyl)- 2,3-dinitro-1,4-butanediol[J].ChineseJStructChem, 2012(3): 415-419.
[6] Chavez D E, Hiskey M A, Naud D L. Synthesis of an energetic nitrate ester[J].Angew.ChemIntEd, 2008, 47: 8307-8314.
[7] Stanislaw C, Marcin N, Artur C, Mateusz S. Synthesis ,Structure and Explosive of a new trinitrate derivative of an unexpected condensation product of nitromethane with glyoxal[J].Propellants,Explosives,Pyrotechnics, 2012, 37: 261-266.
[8] Sheldrick G M.SHELXS-97, Program for Crystal Structure Solution[CP]. University of Göttingen, Germany 1997.
[9]Sheldrick G M.SHELXL-97, Program for Crystal Structure Refinement[CP]. University of Göttingen, Germany 1997.
[10] Smart and Saint. Area Detector Control and Integration Software [CP]. Siemens Analytical X-Ray Systems, Inc. Madison, Wisonsin, USA 1996.
[11] Allen F H, Kennard O, Waston D G. Tables of bond lengths determined by x-ray and neutron diffraction.Part I . Bond lengths in organic compounds[J].JChemSoc,PerkinTransⅡ, 1987,S1-S4.
