
鸭坦布苏病毒竞争定量PCR检测方法的建立与应用
2015-05-06 19:15袁朋等
山东农业科学 2015年3期
袁朋等

摘要: 在现行PCR检测鸭坦布苏病毒(DTMUV)方法的基础上,设计制备DTMUV竞争模板并以其为定量内标物建立竞争定量PCR(QC-PCR)体系。该体系特异性强、灵敏性高,竞争模板和目标模板可在400个分子/μL的水平上共扩增。临床应用结果表明,建立的体系能够满足简单快速确定病毒含量的要求。
关键词:鸭坦布苏病毒;竞争定量PCR;定量检测
中图分类号:S858.32文献标识号:A文章编号:1001-4942(2015)03-0113-05
Establishment and Application of Quantitatively
Competitive PCR System for DTMUV Detection
Yuan Peng1,2*, Wang Bin3*, Xu Chuantian1, Yang Shaohua1, Huang Qinghua1, Zhang Xiumei1, Zhang Lin1**
(1.Institute of Animal Science and Veterinary Medicine, Shandong Academy of Agricultural Sciences/Shandong Key Laboratory of
Animal Diseases Control and Animal Breeding, Jinan 250100, China;2. College of Animal Science and Technology, Shandong Agricultural
University, Taian 271018, China; 3. Department of Bio-Engineering, Henan Institute of Science and Technology, Xinxiang 453003, China)
AbstractBased on the traditional PCR method for DTMUV detection, the competitive template targeting to DTMUV NS5 gene was designed and used as competitor in the quantitatively competitive PCR (QC-PCR) system with high specificity and sensitivity. The target and competitive templates could be co-amplified at the level of 400 molecules per microliter. The clinical application results indicated that the developed QC-PCR could simply, rapidly and quantitatively diagnose the content of DTMUV.
Key wordsDTMUV;QC-PCR;Quantitative detection
目前我国养鸭业迅速发展,肉鸭、蛋鸭存栏量和鸭肉出口量均居世界首位,但近年来随着养殖规模的扩大,鸭病的发生也呈上升趋势,对养鸭业的危害日益严重。2010年,一种新型的鸭源黄病毒——鸭坦布苏病毒(Duck Tembusu Virus, DTMUV)在我国规模化养鸭场爆发并迅速蔓延至全国,给我国养鸭业带来了巨大的经济损失[1,2],据统计,该病仅在2010年造成的经济损失就达50亿元。
DTMUV属黄病毒科、黄病毒属,为有囊膜不分节段单股正链RNA病毒[3]。鸭感染DTMUV后主要出现神经症状、采食量减少、产蛋严重下降甚至绝产,并引起大量死亡。同时感染DTMUV的鸭还容易继发传染性浆膜炎、大肠杆菌病等其他疾病,且疾病发生后由于大量应用药物,导致药物残留严重,影响到鸭肉品质和人类健康。此外,近年来动物流感(H1N1、H7N9、H10N8)相继爆发,病毒多次感染人类,造成死亡,严重危害人类健康,而水禽是流感病毒的重要自然储存宿主。因此,控制鸭病毒性疾病的流行,不仅能够减少养鸭业的经济损失,而且能够促进鸭产业链的健康发展,保障人类健康。
目前尚没有针对性的治疗鸭坦布苏病毒感染的药物,亦缺乏有效的疫苗,因此对病毒进行深入研究、开发预防性和治疗性药物势在必行。但DTMUV活力较差,体外培养稳定性不足[4],这种特性使得其滴度变化快,相关深入研究困难极大,所以找到一种能够快速确定病毒含量且操作便捷的检测方法是后续研究的保证。本研究基于PCR法快速、特异的优点,在现行定性检测方法的基础上[5,6],设计制备DTMUV竞争模板并以其为定量内标物建立竞争定量PCR(Quantitatively Competitive Polymerase Chain Reaction, QC-PCR)检测体系,用于DTMUV的含量测定,为病毒的深入研究及预防控制提供强有力的手段和技术支持。
1材料与方法
1.1毒株
DTMUV分离株、新城疫病毒La Sota株(Newcastle Disease Virus, NDV)、H9N2亚型禽流感病毒(Avian Influenza Virus, AIV)、禽呼肠孤病毒(Avian Reovirus, ARV)、减蛋综合症病毒(Egg Drop Syndrome Virus,EDSV)均由山东省畜禽疫病防治与繁育重点实验室分离、鉴定、保存。
1.2试剂和材料
Ex Taq酶、禽源反转录酶(AMV)、随机引物(9 mer)、TRIzol Reagent购自宝生物工程(大连)有限公司;质粒小提试剂盒、琼脂糖凝胶回收试剂盒、DNA分子量标准(DNA Marker)购自天根生化科技(北京)有限公司;感受态细胞购自北京全式金生物技术有限公司。endprint
1.3试验方法
1.3.1DTMUV的分离与扩增无菌采集疑似DTMUV感染的病变组织,按常规处理方法制成匀浆,反复冻融3次,8 000 r/min离心10 min,弃沉淀,上清加入双抗4℃作用4 h,然后经尿囊腔接种9~11日龄SPF鸡胚,收取尿囊液进行检测和进一步传代培养。
1.3.2病毒RNA提取和cDNA第一链合成按TRIzol Reagents(Invitrogen)试剂盒操作说明提取病毒总RNA,然后进行反转录。反转录体系20 μL,总RNA 2 μL,5×Reverse transcriptase buffer 4 μL,dNTP (2.5 mmol/L)8 μL,RNA抑制剂0.5 μL,random primer(9 mer) 50 pmol,AMV 2 μL,DEPC水补足20 μL。反转录反应条件为30℃ 10 min,42℃ 1 h,99℃ 5 min,4℃ 10 min,即合成cDNA第一链。
1.3.3引物设计与合成参照张琳等[6]建立的巢式PCR检测DTMUV的方法和Celi等[8]构建竞争模板的原理,设计了3条针对DTMUV NS5基因的引物(表1),分别用于目标模板(Object template, 以下简称O)和竞争模板(Competitive template, 以下简称C)的扩增,进而用于可定量的竞争PCR反应。其中P1为共用上游引物,P2为扩增O的下游引物,P4为扩增C的下游引物。引物由生工生物工程(上海)有限公司合成。
1.3.4目标模板、竞争模板的制备分别以P1/P2、P1/P4为上下游引物进行O和C的扩增,PCR扩增体系为25 μL,其中10×Ex Taq buffer 2.5 μL,dNTP(2.5 mmol/L) 2 μL,上下游引物(100 pmol/μL)各0.5 μL,Ex Taq 酶0.5 μL,DTMUV cDNA第一链0.5 μL, ddH2O补足25 μL。反应条件为98℃预变性5 min;94℃变性30 s,47℃退火30 s,72℃延伸40 s, 30个循环;再72℃延伸10 min,4℃终止反应。PCR产物经2%琼脂糖凝胶电泳和紫外观察拍照后,切胶回收,一部分与T载体连接后进行测序;另一部分则于nanodrop上测定浓度,然后根据片段分子量,精确计算其所含分子数,10倍梯度稀释制备不同浓度的O和C。
1.3.5QC-PCR检测体系的建立以P1、P2为上下游引物进行QC-PCR反应,体系25 μL,其中包括10×Ex Taq buffer 2.5 μL,dNTP(2.5 mmol) 2 μL,P1和P2(100 pmol/μL)各0.5 μL,Ex Taq酶0.5 μL,病毒目标模板0.5 μL,竞争模板0.5 μL,ddH2O补足25 μL。反应条件为98℃预变性5 min;94℃变性30 s,47℃退火30 s,72℃延伸40 s, 30个循环;再72℃延伸10 min,4℃终止反应。采用琼脂糖凝胶电泳检测扩增结果。
1.3.6QC-PCR方法的准确性取10倍系列稀释的C分别加入不同反应管中,随后每个反应管中加入同一浓度O进行混合,最后加入QC-PCR反应的其他组分进行共扩增,以验证建立的体系能否使C和O等量共扩增。
1.3.7QC-PCR方法的特异性分别提取DTMUV、NDV、AIV、ARV的RNA和EDSV的DNA,RNA进行反转录得到cDNA第一链。随后分别加入制备的C,用1.3.5中建立的检测体系进行定性和定量共扩增,以检测体系的特异性。
1.3.8QC-PCR方法的灵敏性以1.3.4中制备的10倍系列稀释的O和C为基础,取相同浓度的两种模板等量混合,从而得到一些含相同浓度O+C的反应体系。采用建立的方法进行扩增,以确定两模板可等量扩增的最低浓度。
1.3.9QC-PCR方法的临床应用对送检的发病鸭病料进行无菌采集,加入5倍量PBS(pH 7.2)进行研磨制成匀浆,反复冻融3次,8 000 r/min离心10 min,取上清按TRIzol Reagents(Invitrogen)试剂盒操作说明提取病毒总RNA,反转录后用已建立的竞争定量PCR进行检测。
2结果与分析
2.1目标模板与竞争模板的获得
在前期预试验确定的反应条件下,分别以P1/P2、P1/P4为上下游引物扩增O和C。结果显示,获得了两条约为400 bp和350 bp的特异性条带,分别与O和C大小相符,且测序结果与参考序列一致。将扩增产物进行回收纯化、测定浓度,并计算出每微升分子数分别为4×1013和5.3×1012,将其分别稀释成4×109个分子/μL的初始浓度后10倍系列稀释,制备成4×109~4×102个分子/μL的系列O和C备用。
2.2QC-PCR反应的建立
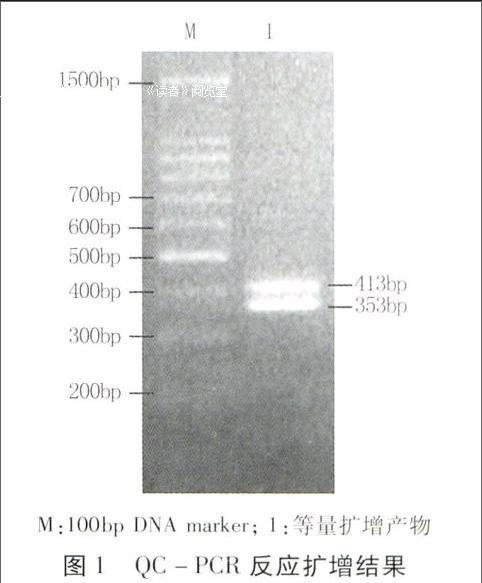
以P1、P2为上下游引物,备用的O和C为反应模板进行竞争定量PCR反应体系的建立。O和C使用相同浓度,经电泳检测(图1)显示,该体系成功扩增353 bp和413 bp的条带,且亮度相当,引物二聚体几乎不存在。表明等量模板在建立的竞争体系中可等量高效扩增。
2.3QC-PCR方法的准确性验证
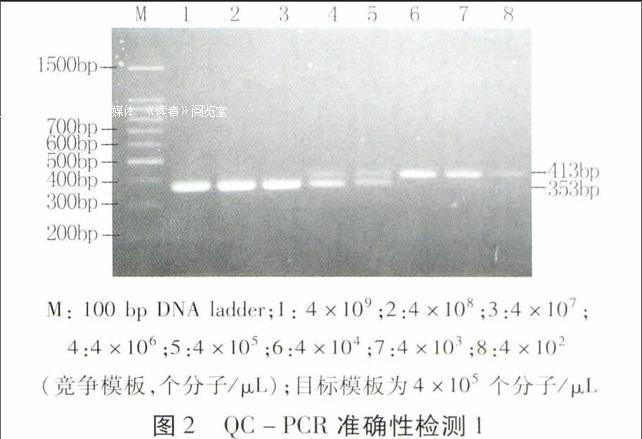
在准确性验证试验中,每个反应由8个子PCR扩增体系组成。在8个反应管中首先分别加入制备的4×109~4×102个分子/μL的系列C各0.5 μL,然后加入同一浓度病毒目标模板,最后加入反应体系的其他组分。在本研究中,选择浓度为4×107个分子/μL和 4×105个分子/μL的O进行准确性验证。结果(图2、图3)表明,两个浓度的模板均能够在建立的QC-PCR体系中与相同浓度的竞争模板等量共扩增,而与其他浓度不能等量共扩增。
2.4QC-PCR方法的特异性endprint
应用建立的QC-PCR反应体系对DTMUV和其他对照病原(NDV、AIV H9N2、ARV、EDSV)在相同条件下进行扩增(体系组成同2.3),以检验体系的特异性。结果显示,对照病原反应组结果相同,仅出现353 bp大小的条带,而没有413 bp扩增产物,表明不含有扩增的目的条带;DTMUV反应组则出现了353 bp和413 bp的条带,且两条带在浓度为4×105个分子/μL时亮度相当(图4),所以DTMUV的病毒样品浓度大概为4×105个分子/μL。
2.6QC-PCR对临床样品的检测
提取15份送检病料的核酸,应用建立的QC-PCR体系进行检测,其中9份为阳性,且病毒检测量大致在4×103~4×107个分子/μL之间不等。
3讨论
由于DTMUV活力较差,56℃加热15 min即灭活,即使在37℃的环境中,24 h后病毒滴度下降也非常明显,所以病毒含量是时刻变化的。基于DTMUV的不稳定特性,在各项研究前进行病毒含量的测定是十分必要的。虽然目前应用于DTMUV的快速诊断方法较多,如RT-PCR[5]、巢式PCR[6]、RT-LAMP、荧光定量PCR、血清学方法[7]等,且检测效果良好,但这些方法在满足操作简单、成本低廉、可定量检测要求时表现出了极大的局限性:PCR和LAMP反应有很高的敏感性和操作性,但不能够对扩增产物进行定量;荧光定量PCR能够定量,但仪器昂贵、成本高;而血清学检测方法试验要求较高,操作复杂。本研究建立的QC-PCR,一方面可以代替普通PCR反应进行定性反应,鉴定样品中是否含有目的核酸;另一方面还可以进行定量反应,将几个普通的PCR反应进行组合,加入制备的竞争模板即可对样品进行定量,以满足对病毒降解状况的了解。同时,建立的QC-PCR反应体系具有很高的敏感性和特异性。
建立QC-PCR反应体系的关键是竞争模板(亦称竞争子,Competitor)的制备。DTMUV NS5基因在所有片段中保守性最高,且种间差异较大,最适合进行DTMUV的定量检测体系引物的设计。竞争定量模板构建的方法较多[8~12],本研究采用Celi等构建竞争模板的原理,针对DTMUV的NS5基因设计了3条引物,成功地扩增得到比目标模板少60 bp的竞争模板。经过准确性和敏感性试验,证明两种模板在浓度相同时扩增效率相近,符合QC-PCR的要求。
本研究建立的QC-PCR方法敏感性高、特异性强、成本低廉、操作简便,重要的是能够基于DTMUV的特性进行定量检测,为研究人员明确DTMUV活力状态、增殖特性及进行后续的深入研究提供了强有力的手段。
参考文献:
[1]张大丙.鸭出血性卵巢炎的研究进展[J].中国家禽,2011, 33 (14) : 37-38.
[2]曹贞贞,张存,黄瑜,等. 鸭出血性卵巢炎的初步研究[J]. 中国兽医杂志, 2010, 46(12): 3-6.
[3]Leyssen P, De Clercg E, Neyts J. Perspectives for the treatment of infections with flaviviridae[J]. Clin. Microbiol. Rev., 2000, 13(1): 67-82.
[4]马秀丽, 于可响,高凤,等. 一株鸭黄病毒的生物学特性研究[C]//山东畜牧兽医学会禽病学专业委员会第二届禽病学术研讨会. 2011: 221-224.
[5]张帅,云涛,叶伟成,等.鸭坦布苏病毒一步法RT-PCR检测方法的建立和应用[J]. 浙江农业学报,2012,24(1):37-40.
[6]张琳, 胡北侠, 颜世敢, 等. 鸭黄病毒巢式PCR检测方法的建立和应用[J]. 福建农业学报, 2012, 27(2): 124-129.
[7]谢星星,李祥瑞,李银,等.检测坦布苏病毒抗体NS1-ELISA方法的建立与初步应用[J]. 浙江农业学报,2014,26(1):135-140.
[8]Celi F S, Zenilman M E, Shuldiner A R. Rapid and versatile method to synthesize internal standards for competitive PCR[J]. Nucleic Acids Research, 1993, 21(4): 1047.
[9]Ballagi-Pordány A, Belak S. The use of mimics as internal standards to avoid false negatives in diagnostic PCR[J]. Molecular and Cellular Probes, 1996,10:159-164.
[10]Jin C F, Mata M, Fink D J. Rapid construction of deleted DNA fragments for use as internal standards in competitive PCR[J].PCR Methods and Applications, 1994(3): 252-255.
[11]Siebert P D, Larrick J W. PCR MIMICS:Competitive DNA fragments for use as internal standards in quantitative PCR[J].Biotechniques, 1993, 14: 244-249.
[12]Ishibashi O. A new method to synthesize competitor RNAs for accurate analyses by competitive RT-PCR[J].Journal of Biochemical Biophysical Methods, 1997, 35: 203-207.endprint
