
多重PCR快速检测两种致泻性大肠杆菌方法的建立
2015-05-05 12:10:23胡文忠何煜波姜爱丽李晓博
食品工业科技 2015年21期
董 妍,胡文忠,何煜波,姜爱丽,冯 可,李晓博
(1.大连工业大学食品学院,辽宁大连 116600;2.大连民族学院生命科学学院,辽宁大连 116600;3.大连理工大学生命科学与技术学院,辽宁大连 116024)
多重PCR快速检测两种致泻性大肠杆菌方法的建立
董 妍1,胡文忠2,*,何煜波2,姜爱丽2,冯 可3,李晓博2
(1.大连工业大学食品学院,辽宁大连 116600;2.大连民族学院生命科学学院,辽宁大连 116600;3.大连理工大学生命科学与技术学院,辽宁大连 116024)
本研究以肠出血性大肠杆菌O157∶H7及肠侵袭性大肠杆菌为目标菌,针对大肠杆菌共同基因uidA、肠出血性大肠杆菌O157∶H7的特异基因O-antigen、肠侵袭性大肠杆菌的特异基因ipaH设计3对引物,建立并优化了检测两种菌的多重PCR检测方法,进行了特异性验证和灵敏度分析,并应用于人工接种新鲜莴苣的检测中。实验结果表明,本研究建立的三重PCR快速检测两种致病菌的检出限为6.3×103CFU/mL,具有较好的灵敏度和特异性。利用所建立的多重PCR方法对人工接种肠出血性大肠杆菌O157∶H7和肠侵袭性大肠杆菌的新鲜莴苣进行检测,检出限为7.8×104CFU/mL。此方法能够对肠出血性大肠杆菌O157∶H7和肠侵袭性大肠杆菌两种食源性致病菌进行快速检测。
多重PCR,快速检测,肠出血性大肠杆菌O157∶H7,肠侵袭性大肠杆菌
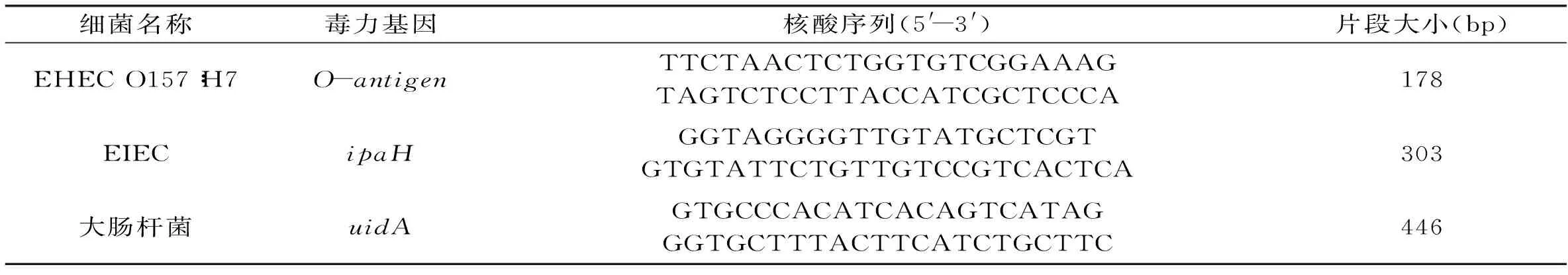
表1 多重PCR引物序列
大肠杆菌(Escherichiacoli,E.coli)作为人和动物肠道内的优势菌,大部分不具有致病性,但是少部分血清型的大肠杆菌可以引起人体以腹泻症状为主的疾病,严重者可危及生命。根据不同血清型的致泻大肠杆菌的生物学特性,国际上将其分为肠产毒性大肠杆菌(EnterotoxigenicE.coli,ETEC)、肠致病性大肠杆菌(EnteropathogenicE.coli,EPEC)、肠出血性大肠杆菌(EnterohemorrhagicE.coli,EHEC)、肠侵袭性大肠杆菌(EnteroinvasiveE.coli,EIEC)和肠粘附性大肠杆菌(EnteroaggregativeE.coli,EAEC)五大类[1]。致泻大肠杆菌可引起食源性疾病的传播与暴发,在我国是主要的食源性致病菌之一,因此,针对不同血清型的致泻大肠杆菌研究快速、特异、灵敏的检测方法,对保障食品安全及人们生命健康有着重要意义。
肠出血性大肠杆菌O157∶H7是肠出血性大肠杆菌最常见的血清型,具有致病性强、死亡率高的特点,并且其感染剂量极低,感染10~100个活菌即可致病;EIEC可引起发热、剧烈腹痛、严重水样腹泻、血性腹泻等症状,是引起痢疾样腹泻的重要食源性致病菌之一[2]。因此,快速、特异、灵敏地检测两种致泻性大肠杆菌对有效控制食源性致病菌的传播、预防食源性疾病的暴发至关重要。
检测不同血清型的致泻大肠杆菌常用传统检测方法,但它存在着操作复杂、检测周期长、检测单一、检测成本高等不足,并且食品中的致病菌往往数量较少,应用传统检测方法会造成漏检的结果,越来越不能满足人们对食品质量与安全的要求[3]。随着食源性致病菌检测技术的高速发展,分子生物学因高效、灵敏、准确,普遍应用于食源性致病菌检测中。特别是多重PCR技术,能够同时检测多种致病菌,与实时荧光定量PCR等其他分子生物学技术相比,提高检测效率的同时也降低了检测成本,具有广阔的发展前景而成为研究热点之一[4]。
本研究针对EHEC O157∶H7和EIEC的毒力基因和大肠杆菌的共有基因,设计了三对特异性引物,建立了同时检测EHEC O157∶H7和EIEC的多重PCR方法,以期为快速检测食品中不同血清型致泻性大肠杆菌提供参考。
1 材料与方法
1.1 材料与仪器
菌株 肠出血性大肠杆菌标准菌株O157∶H7 CICC 21530、肠侵袭性大肠杆菌标准菌株CICC 10661、肠产毒性大肠杆菌标准菌株CICC 10414、肠致病性大肠杆菌标准菌株CICC 10372、大肠杆菌标准菌株CMCC 44102、鼠伤寒沙门氏菌(Salmonellatyphimurium)标准菌株ATCC 14028、乙型副伤寒沙门氏菌(SalmonellaparatyphiB)标准菌株CMCC 50094、三株单核细胞增多性李斯特菌(Listeriamonocytogenes)标准菌株ATCC 19111、ATCC 19112、ATCC 19115、金黄色葡萄球菌(Staphylococcusaureus)标准菌株ATCC 6538、副溶血性弧菌(VibrioParahemolyticus)标准菌株CICC 10435,均由大连民族学院生命科学学院食品安全实验室提供。
试剂 LB营养琼脂培养基、LB营养肉汤培养基 由青岛海博生物技术有限公司生产;乳糖、甘露醇等为国产分析纯试剂;琼脂糖由BIOWEST公司生产,GelRed染料由Biotium公司生产;10×PCR Buffer(Mg2+free)、细菌DNA提取试剂盒等由宝生物工程(大连)有限公司生产。
DYCP-31F型电泳仪 由北京市六一仪器厂生产;BioSpectrum 310 凝胶成像系统由美国UVP公司生产;Thermo Arktic PCR仪 由Thermo Fisher Scientific公司生产;全自动高压灭菌锅由SANYO公司生产。
1.2 实验方法
1.2.1 引物的设计与合成 针对EHEC O157∶H7的毒力基因O-antigen[5]、EIEC的毒力基因ipaH[6],以及大肠杆菌共同的基因uidA[7],根据GenBank中公布的相关序列,应用软件Primer Premier 5.0和Oligo 6.0设计3对特异性引物[8],引物序列见表1,由宝生物工程(大连)有限公司合成。
1.2.2 菌悬液和DNA模板的制备 将两种致泻大肠杆菌的标准菌株分别接种于LB营养肉汤培养基中,培养基中已加入了0.3 g/100 mL的甘露醇和1.0%的乳糖[9],37 ℃振荡培养过夜,得到菌悬液。取菌悬液1 mL,用细菌DNA提取试剂盒提取细菌DNA,-20 ℃保存备用。
1.2.3 单重PCR反应的建立 分别以EHEC O157∶H7和EIEC的基因组DNA为模板。反应体系为25 μL:10×PCR Buffer(Mg2+free)2.5 μL,MgCl2(25 mmol/L)1.5 μL,dNTP Mixture(2.5 mmol/L each)2 μL,DNA模板(EHEC O157∶H7的DNA模板浓度为16.8 ng/μL;EIEC的DNA模板浓度为10.75 ng/μL)2.5 μL,引物(4 μmol/L)各1 μL,rTaq酶(5 U/μL)0.2 μL,加RNase-Free Water至25 μL;在反应中设置梯度退火温度以明确最佳退火温度,依次为51.0、51.6、52.4、53.4、54.6、56.1、57.5、58.5、59.3、60 ℃,PCR反应条件为94 ℃预变性4 min,94 ℃ 30 s、梯度退火温度30 s、72 ℃ 40 s,34个循环,72 ℃延伸7 min[10]。扩增产物用2%琼脂糖凝胶电泳检测,95 v电泳1 h,在凝胶成像系统下观察结果[11]。
1.2.4 多重PCR 反应的建立 多重PCR反应体系为25 μL:10×PCR Buffer(Mg2+free)2.5 μL,MgCl2(25 mmol/L)1.5 μL,dNTP Mixture(2.5 mmol/L each)2 μL,DNA模板(EHEC O157∶H7的DNA模板浓度为16.8 ng/μL;EIEC的DNA模板浓度为10.75 ng/μL)各1 μL,引物(4 μmol/L)各1 μL,rTaq酶(5 U/μL)0.2 μL,加RNase-Free Water至25 μL。按建立的单重PCR反应条件进行扩增。
1.2.5 多重PCR反应的优化 Mg2+(25 mmol/L)添加量的优化:反应体系中Mg2+的添加量依次为1.0、1.5、2.0、2.5、3.0、3.5、4.0 μL;rTaq酶(5 U/μL)添加量的优化:反应体系中rTaq酶的添加量依次为0.1、0.2、0.3、0.4、0.5、0.6 μL;引物(4 μmol/L)添加量的优化:在合理的引物终浓度范围内选择0.12、0.16、0.2 μmol/L三个水平,以0.04 μmol/L的浓度梯度递增,折合为在反应体系内应添加的体积,通过L9(33)正交设计表(表2)进行实验;退火温度的优化:退火温度依次设置为51.0、51.6、52.4、53.4、54.6、56.1、57.5、58.5、59.3、60 ℃。根据各项优化结果选出体系中Mg2+、rTaq酶、引物的最适添加量及最适的退火温度。
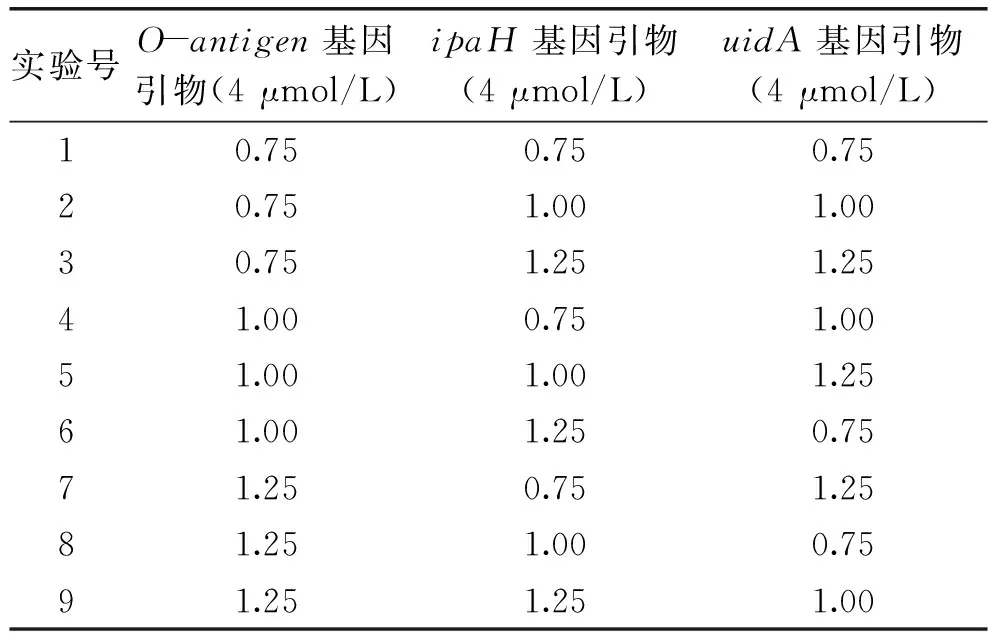
表2 引物添加量优化实验正交设计表
注:添加量单位为μL。
1.2.6 多重PCR反应特异性验证 为验证上述建立的多重PCR方法的准确性,按建立好的多重PCR反应,每组按照表3添加模板DNA和引物,进行扩增反应。同时以EHEC O157∶H7标准菌株CICC 21530、EIEC标准菌株CICC 10661、ETEC标准菌株CICC 10414、EPEC标准菌株CICC 10372、大肠杆菌标准菌株CMCC 44102、鼠伤寒沙门氏菌标准菌株ATCC 14028、乙型副伤寒沙门氏菌标准菌株CMCC 50094、三株单增李斯特菌标准菌株ATCC 19111、ATCC 19112、ATCC 19115、金黄色葡萄球菌标准菌株ATCC 6538、副溶血性弧菌标准菌株CICC 10435的DNA为模板,对引物进行特异性验证,并用RNase-Free Water作空白对照。
1.2.7 多重PCR反应检出限检测 将菌悬液分别用无菌生理盐水按10倍梯度稀释至10-6,取1 mL用细菌DNA提取试剂盒提取模板DNA,按最佳反应体系与反应条件进行扩增。同时,两种菌各取10-5、10-6、10-7三个稀释梯度按照涂布法进行菌落计数,以确定细菌的浓度。
1.2.8 应用多重PCR方法检测人工模拟样品 将新鲜莴苣用无菌水洗净后在75%的酒精中浸泡10 min,取出在无菌操作下取出置于无菌的生物安全柜中通风至酒精完全挥发,此时莴苣样品表面干燥,无酒精液滴存在,此过程要将莴苣样品尽量平铺以获得尽可能大的挥发面积。称取10 g,加入到90 mL已添加了0.3 g/100mL的甘露醇和1.0%的乳糖的LB营养肉汤培养基中,取菌悬液1 mL加入,37 ℃过夜培养。培养后均质,将均质液用无菌生理盐水按10倍梯度稀释至10-6,取1 mL用细菌DNA提取试剂盒提取模板DNA,按最佳反应体系与反应条件进行扩增。同时,两种菌各取10-5、10-6、10-7三个稀释梯度按照涂布法进行菌落计数,以确定样品中细菌的浓度。

表3 准确性验证实验
注:“+”代表添加;“-”代表不添加。
2 结果与分析
2.1 单重PCR反应的建立
研究中对各个引物的退火温度进行了选择(图1),结果显示EHEC O157∶H7和EIEC在所有温度梯度均可以扩增出目标条带,且扩增条带的亮度几乎无差异,而大肠杆菌的通用引物随着退火温度的增加,扩增出的条带逐渐暗淡,在51.0~54.6 ℃的温度范围内,扩增出的条带都清晰明亮,说明在此范围内的温度均能够高效扩增,满足实验要求,因此选择53 ℃为单重PCR扩增的退火温度。建立的单重PCR反应见图2,EHEC O157∶H7在178 bp处出现目的条带,EIEC在303 bp处出现目的条带,大肠杆菌通用引物对两种致泻大肠杆菌模板DNA分别扩增,都在446 bp处出现的目的条带,目的条带与预期大小符合。
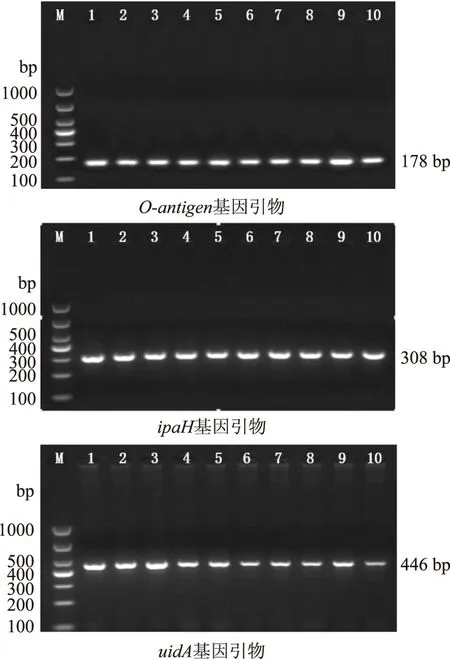
图1 单重PCR反应退火温度的选择Fig.1 Optimization of single PCR annealing temperature注:M:Marker;1~10:退火温度分别为51.0、51.6、52.4、53.4、54.6、56.1、57.5、58.5、59.3、60 ℃。
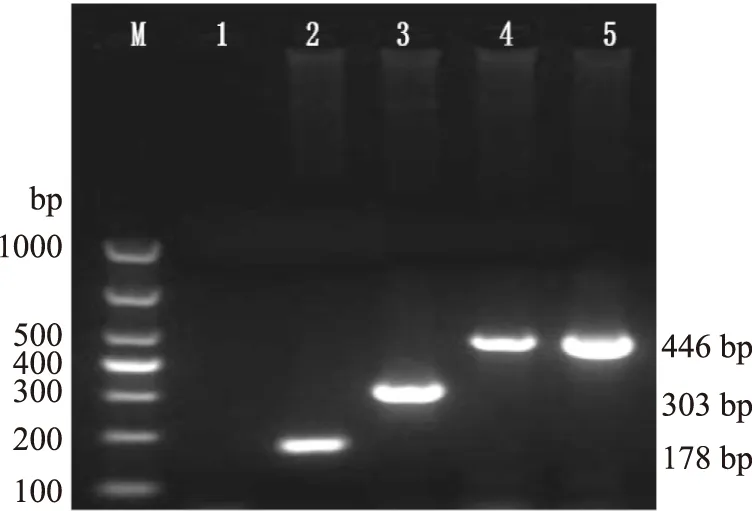
图2 单重PCR反应的建立Fig.2 Establishment of single PCR注:M:Marker;1:阴性对照;2:EHEC O157∶H7(O-antigen基因);3:EIEC(ipaH基因);4:EHEC O157∶H7(uidA基因);5:EIEC(uidA基因)。
2.2 多重PCR反应的建立
多重PCR反应结果显示(图3),对EHEC O157∶H7的O-antigen基因和uidA基因进行双重扩增反应,在178 bp和446 bp处出现两条特异性目的条带;对EIEC的ipaH基因和uidA基因进行双重扩增反应,在303 bp和446 bp处出现两条特异性目的条带;将两种致泻大肠杆菌组合后对三种目的基因进行多重PCR反应,在178、303、446 bp处出现三条特异性目的条带。目的条带与预期大小符合,条带清晰且无非特异性扩增。
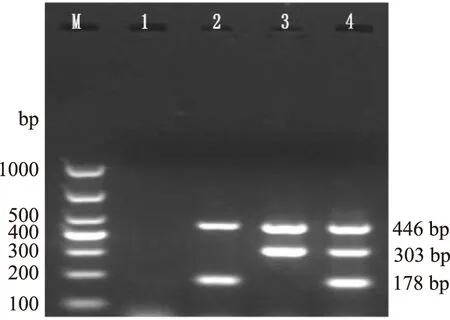
图3 多重PCR反应的建立Fig.3 Establishment of multiplex PCR注:M:Marker;1:阴性对照;2:EHEC O157∶H7;3:EIEC;4:EHEC O157∶H7和EIEC。
2.3 多重PCR反应优化结果
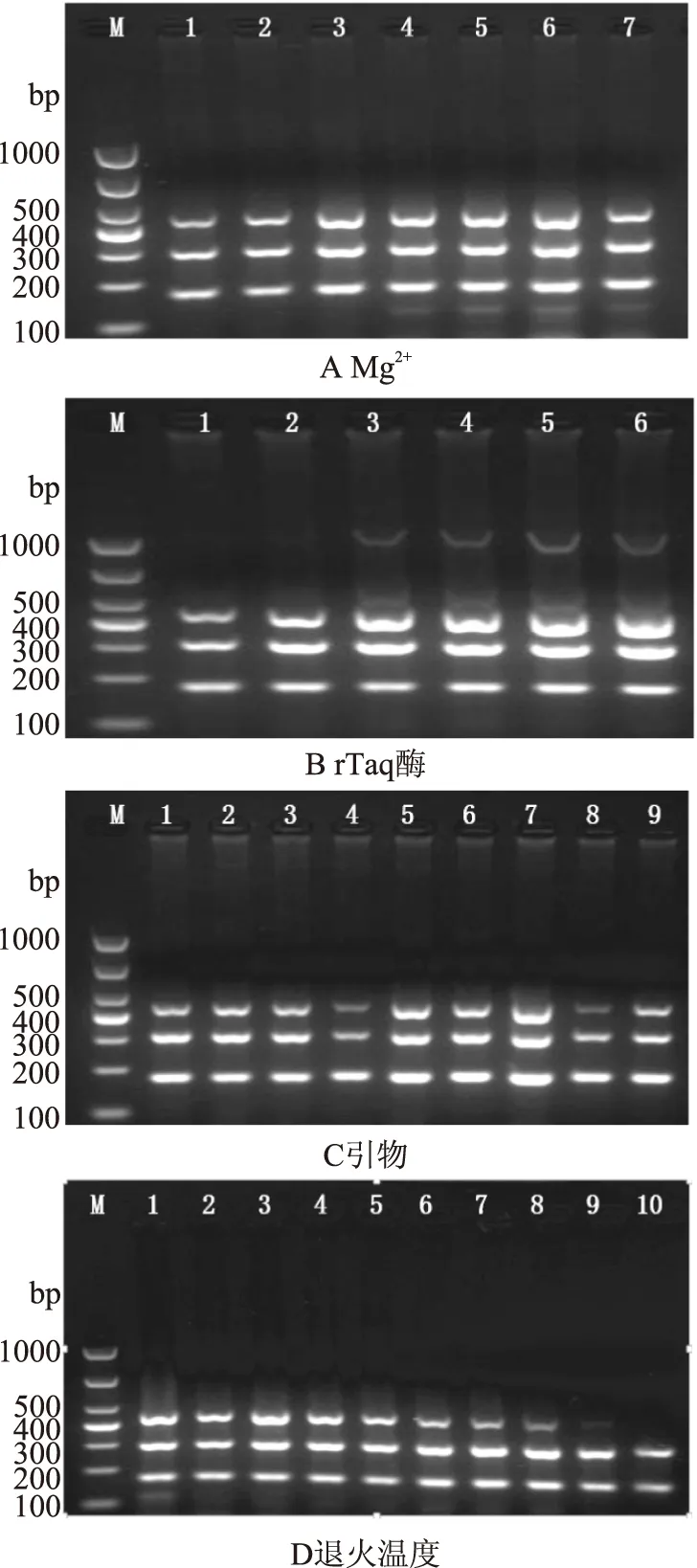
图4 多重PCR反应优化结果Fig.4 The optimization results of multiplex PCR注:图A(M:Marker;1~7:Mg2+添加量分别为1.0、1.5、2.0、2.5、3.0、3.5、4.0 μL);图B(M:Marker;1~6:rTaq酶添加量分别为0.1、0.2、0.3、0.4、0.5、0.6 μL);图C(M:Marker;1~9:引物添加量见表2);图D(M:Marker;1~10:退火温度分别为51.0、51.6、52.4、53.4、54.6、56.1、57.5、58.5、59.3、60 ℃)。
对多重PCR反应优化的结果见图4。不同的Mg2+(25 mmol/L)添加量均可以扩增出目的条带,添加量为2.5~4.0 μL时存在非特异性扩增条带,根据扩增条带的明暗程度,Mg2+的添加量选择2.0 μL。rTaq酶(5 U/μL)添加量为0.3~0.6 μL时存在非特异性扩增条带,根据目标条带的明暗,rTaq酶添加量选择0.2 μL。通过正交实验对引物(4 μmol/L)的添加量进行优化的结果显示,在所有引物添加组别中均未出现非特异性扩增条带,当引物添加组别为第5组、第6组、第7组时,扩增出的目标条带较明亮,进一步比较三个组别,第6组扩增出的三条目标条带的明暗程度相差较大,uidA基因引物扩增出的条带亮度明显微弱于其余两条条带,不适合在多重PCR反应中应用,第7组扩增出的三条条带存在拖尾的现象,并且存在不同程度的条带下移现象,与预期片段长度稍有偏差,特别是uidA基因引物扩增出的条带,拖尾和下移情况明显,不适合在多重PCR反应中应用,因此引物添加量选择的组别为第5组,具体添加量为:O-antigen基因的引物为1 μL,ipaH基因的引物为1 μL,uidA基因的引物为1.25 μL。
对退火温度进行优化的结果显示,在51 ℃时存在非特异性扩增,在温度为51.6~53.3 ℃的范围内,扩增出的目标条带最明亮,说明此范围内的温度均可以满足实验要求,从54.6 ℃开始,扩增条带越来越暗淡,表现最明显的是uid基因引物扩增出的目标条带,在59.3 ℃以后,不能扩增出目标条带,适当提高退火温度可以避免非特异扩增的产生,并且考虑到PCR仪存在着不可避免的温度误差,因此在51.6~53.3 ℃的温度范围内,退火温度选择53 ℃即可满足实验要求。
最终建立的多重PCR反应体系为:10×PCR Buffer(Mg2+free)2.5 μL,MgCl2(25 mmol/L)2 μL,dNTP Mixture(2.5 mmol/L)2 μL,DNA模板各1 μL,O-antigen基因引物(4 μmol/L)各1 μL,ipaH基因引物各1 μL,uidA基因引物各1.25 μL,rTaqDNA聚合酶(5 U/μL)0.2 μL,加RNase-Free Water至25 μL。多重PCR反应条件为94 ℃预变性4 min,94 ℃ 30 s、53 ℃ 30 s、72 ℃ 40 s,34个循环,72 ℃延伸7 min。
2. 4 多重PCR反应特异性验证
对建立的多重PCR反应的准确性进行验证时,通过对不同组合的模板DNA和引物进行扩增反应,均获得了预期的扩增条带(图5),没有非特异性扩增条带产生;用所建立的多重PCR 方法进行特异性检测,结果显示(图6),O-antigen基因引物只能特异性扩增EHEC O157∶H7,ipaH基因引物只能特异性扩增EIEC,uidA基因引物能够扩增出所有种类的大肠杆菌,包括EHEC O157∶H7、EIEC、ETEC、EPEC和大肠杆菌CMCC 44102,而其他菌扩增结果均为阴性,说明所建立的方法特异性良好。
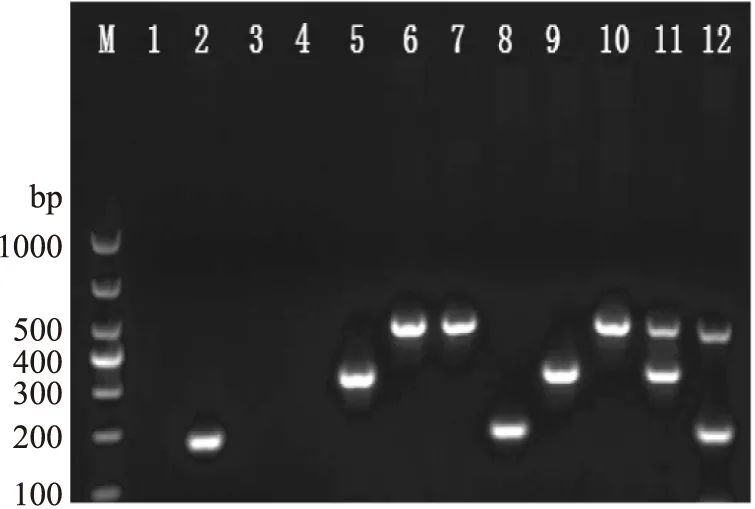
图5 多重PCR反应准确性验证结果Fig.5 The results of accuracy verification 注:M:Marker;1~12:与表3组号一致。
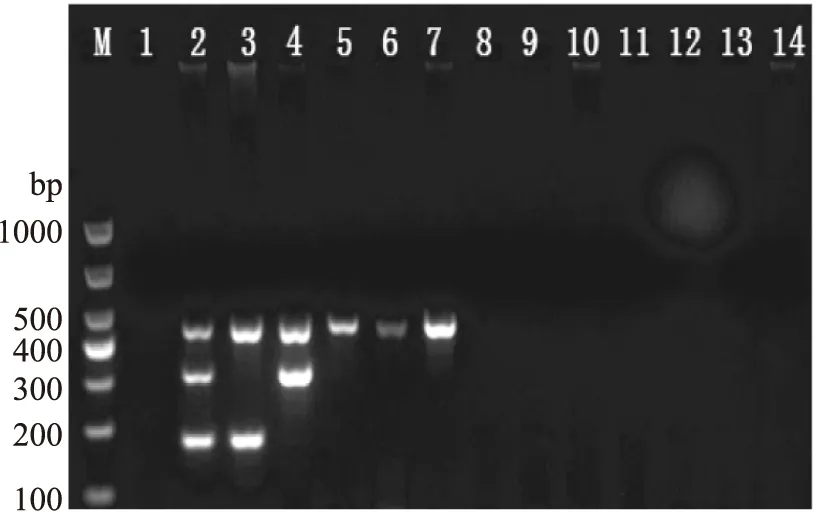
图6 多重PCR反应特异性验证结果Fig.6 The specificity of multiple PCR注:M:Marker;1:阴性对照;2:阳性对照;3:EHEC O157∶H7;4:EIEC;5:ETEC;6:EPEC;7:大肠杆菌CMCC 44102;8:鼠伤寒沙门氏菌;9:乙型副伤寒沙门氏菌;10~12:单增李斯特;13:金黄色葡萄球菌;14:副溶血性弧菌。
2. 5 多重PCR反应灵敏度检测
应用所建立的多重PCR反应对梯度浓度的菌液进行扩增(图7),结合菌落计数,结果显示,菌液从6.3×107CFU/mL稀释至6.3×103CFU/mL时,有扩增条带出现,因此此方法同时检测EHEC O157∶H7和EIEC的检出限为6.3×103CFU/mL。
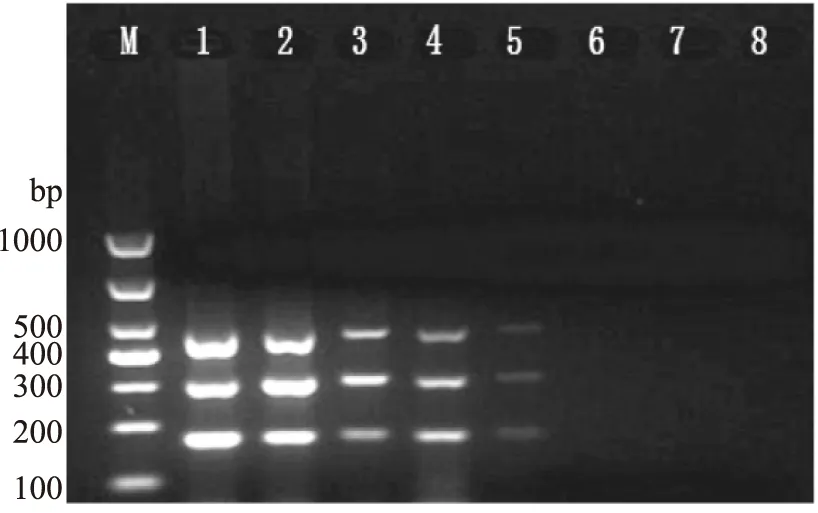
图7 多重PCR反应检测2种致泻大肠杆菌的检出限Fig.7 The sensitivity of detection of two Diarrheagenic Escherichia coli注:M:Marker;1:6.3×107 CFU/mL;2:6.3×106 CFU/mL;3:6.3×105 CFU/mL;4:6.3×104 CFU/mL;5:6.3×103 CFU/mL;6:6.3×102 CFU/mL;7:6.3×101 CFU/mL;8:阴性对照。
2.6 人工模拟样品的多重PCR检测
人工污染的新鲜莴苣过夜培养得到的均质液浓度为7.8×107CFU/mL,经10倍梯度稀释后提取细菌DNA,应用本研究建立的多重PCR方法进行检测后,结果显示(图8),检出限为7.8×104CFU/mL。
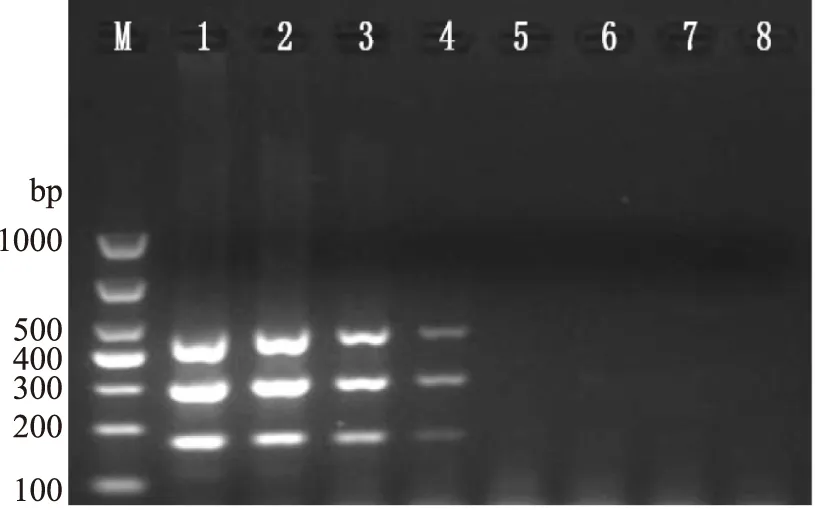
图8 人工污染莴苣样品的检出限检测结果Fig.8 The sensitivity of detection of artificial lettuce infected with two pathogens注:M:Marker;1:7.8×107 CFU/mL;2:7.8×106 CFU/mL;3:7.8×105 CFU/mL;4:7.8×104 CFU/mL;5:7.8×103 CFU/mL;6:7.8×102 CFU/mL;7:7.8×101 CFU/mL;8:阴性对照。
3 结论与讨论
在目前的研究中,应用多重PCR检测技术快速检测不同种类食源性致病菌的研究较广泛[12-14],但是针对不同血清型的致泻性大肠杆菌的快速检测技术研究较匮乏,并且由于不同血清型致泻性大肠杆菌的培养特性、生化特性相似,抗原性多有交叉,因此从分子的角度对不同血清型致泻性大肠杆菌进行鉴定无疑是最可靠、最有效的检测手段之一。张铁男[15]等人对ETEC、EPEC、EIEC三种致泻性大肠杆菌应用多重PCR方法进行检测;潘劲草[16]等人应用多重PCR方法对EHEC和EPEC的多种毒力基因进行了检测。
然而,在多重PCR反应中存在着许多影响因素,各引物的特异性、稳定性就是影响因素之一,因此合理设计引物、优化反应体系中不同引物浓度就十分重要。本研究针对两种致泻大肠埃希氏菌特异的毒力基因和大肠杆菌共有的毒力基因,设计了三对引物,在反应中由O-antigen基因和uidA基因的引物共同鉴定EHEC O157∶H7,由ipaH基因和uidA基因的引物共同鉴定EIEC,避免了将此两种菌错误鉴定为其他的食源性致病菌,特别是其他血清型的致泻大肠杆菌。本研究还对引物添加量进行了正交优化,使不同的引物组合更加全面,得到的最适引物添加量组合更加合理,避免了因引物浓度过高产生的错配及非特异性扩增,也避免了因引物浓度过低使扩增产物太少而造成的假阴性。贺晨[17]等人应用正交实验对多重PCR反应体系进行了优化,多重PCR反应效果良好。另外,本研究还对建立的多重PCR反应进行了准确性验证,旨在验证引物的稳定性和特异性,避免因反应内部产生错配而引起的非特异性扩增、假阳性和假阴性现象,结果证明引物稳定性和特异性良好。
过高的Mg2+浓度和rTaq酶浓度、过低的退火温度都会加大非特异性扩增产生的几率[18],因此本研究选择这三项对多重PCR反应影响较大的因素进行优化实验,能够使所建立的多重PCR反应以最适的反应体系和反应条件对两种致泻性大肠杆菌进行检测。此外,在前期实验中,对延伸时间和循环数也进行了优化,发现这两项因素对本研究基本没有影响,因此在反应条件中只选择对退火温度进行优化。
本研究建立的检测EHEC O157∶H7和EIEC的多重PCR方法检出限为6.3×103CFU/mL,说明建立的方法灵敏度良好,这个结果与贺晨[17]、孟庆美[19]和Rachel Binet[6]等建立的多重PCR检测方法灵敏度接近,高于赵红庆[20]和舒畅[21]等人建立的多重PCR方法的灵敏度,低于董伯森[18]等人将FTA 滤膜法与多重PCR方法结合检测致病菌的检出限。这可能是由于不同方法所应用的DNA提取方法不同、不同细菌模板DNA或引物之间相互抑制作用、扩增反应体系与反应条件的参数不同等原因,造成了灵敏度上的差异。
影响多重PCR反应的因素之一还有细菌的培养环境、模板DNA的含量和质量等,翁思聪[22]等人应用复合型増菌培养基,减少了培养基对PCR检测的影响;郝玉芹[23]等人应用FTA滤膜提取模板DNA,有效提高了灵敏度。本研究中检测人工模拟样品的检出限高于只检测标准菌株的检出限,是由于果蔬中的成分会对细菌模板DNA的提取造成干扰,使模板DNA的含量、纯度等指标降低,从而影响多重PCR反应[21],因此在下一步研究中,应将重心放在对样品的增菌培养、菌体富集或模板DNA提取方法的研究中,以获得特异性强、灵敏度高、针对性强的多重PCR检测技术。
[1]王乃福,吴冬雪,张霞,等.致泻性大肠杆菌基因芯片检测方法的建立[J].食品研究与开发,2014,35(7):5-9.
[2]蒋原.食源性病原微生物检测指南[M].北京:中国标准出版社,2010.77-79.
[3]徐君怡,曹际娟,郑秋月,等.变性高效液相色谱检测食品中致泻性大肠杆菌[J].微生物学报,2008,48(11):1526-1531.
[4]Fakruddin M D,Sultana M,Ahmed M M,et al. Multiplex PCR(polymerase chain reaction)assay for detection ofE.coliO157∶H7,Salmonellasp.,Vibrio cholerae and Vibrio parahaemolyticus in spiked shrimps(Penaeus monodon)[J]. Pakistan journal of biological sciences:PJBS,2013,16(6):267-274.
[5]李睿,张忠美,戴诗皎,等.食品中大肠杆菌O157的PCR检测与wzy基因的测序分析[J].食品科学,2010,31(12):193-196.
[6]Binet R,Deer D M,Uhlfelder S J. Rapid detection of Shigella and EnteroinvasiveEscherichiacoliin produce enrichments by a conventional multiplex PCR assay[J]. Food Microbiology,2014,40(2):48-54.
[7]郝江燕,胡文忠,何煜波,等.鲜活农产品中大肠杆菌的PCR检测[J].食品工业科技,2013,34(17):150-153.
[8]刘志杰,李如举,曾智勇,等.多重PCR反应的影响因素及其优化[J].黑龙江畜牧兽医,2011(7):26-28.
[9]吕晓萌,胡文忠,冯叙桥,等.PCR法检测大肠杆菌的增殖条件优化研究[J].食品工业科技,2014,35(17):134-136.
[10]王慧,朱瑞良,谭燕玲,等.多重PCR检测三种重要食源性致病菌方法的建立及应用[J].中国农业科学,2011,44(11):2334-2340.
[11]吴建英,宋建新,曹金萍,等.多重PCR快速检测3种食源性致病菌[J].实验与检验医学,2014,32(2):146-149.
[12]Son I,Binet R,Maounounen-Laasri A,et al. Detection of five Shiga toxin-producingEscherichiacoligenes with multiplex PCR[J]. Food Microbiology,2014,40(3):31-40.
[13]Gordillo R,Rodríguez A,Werning M L,et al. Quantification of viable Escherichia coli O157∶H7 in meat products by duplex real-time PCR assays[J]. Meat Science,2014,96(2):964-970.
[14]Kumar A,Grover S,Kumar Batish V. Application of multiplex PCR assay based on uidR and fliCH7 genes for detection ofEscherichiacoliO157∶H7 in milk[J]. The Journal of general and applied microbiology,2013,59(1):11-19.
[15]张铁男,李继昌,鲁成武,等.3种致泻性大肠埃希氏菌多重PCR检测方法的研究[J].中国兽医杂志,2007,43(9):17-19.
[16]潘劲草,邵浙新.多重PCR方法检测EHEC和EPEC毒力基因[J].中国人兽共患病杂志,2001,17(1):52-55.
[17]贺晨,孙鸿燕,邵丽筠,等. 4种病原菌多重PCR检测方法建立[J].中国公共卫生,2011,27(4):525-527.
[18]董伯森,杨国兴,井官军,等.利用多重 PCR 检测3种食源性致病菌[J].医学动物防制,2014,30(8):41.
[19]孟庆美,王少辉,韩先干,等.禽致病性大肠杆菌毒力基因多重PCR方法的建立和应用[J].微生物学报,2014,54(6):696-702.
[20]赵红庆,苑锡铜,王玉飞,等.多重PCR检测致腹泻大肠埃希菌方法的建立[J].中国卫生检验杂志,2009,19(10):2223-2226.
[21]舒畅,姜琛璐,钟慈平,等.三种食源性致病菌多重PCR检测方法的建立[J].食品工业科技,2014,35(12):49-54.
[22]翁思聪,朱军莉,冯立芳,等. 1种选择性富集沙门氏菌,志贺氏菌,金黄色葡萄球菌和副溶血性弧菌共增菌培养基SSSV研究[J].中国食品学报,2013,13(1):19-28.
[23]郝玉芹,孙皆宜,李艾,等. 正交优化多重PCR反应体系检测3种食源性致病菌的研究[J].安徽农业科学,2010,38(2):602-605.
Establishment of a multiplex PCR method for rapid detection of two kinds of diarrheagenicEscherichiacoli
DONG Yan1,HU Wen-zhong2,*,HE Yu-bo2,JIANG Ai-li2,FENG Ke3,LI Xiao-bo2
(1.College of Food Engineering,Dalian Polytechnic University,Dalian 116034,China;2.College of Life Science,Dalian Nationalities University,Dalian 116600,China;3.College of Life Science and Biotechnology,Dalian University of Technology,Dalian 116024,China)
To establish a rapid multiplex PCR method,which can simultaneously detectEnterohemorrhagicEscherichiacoliO157∶H7 andEnteroinvasiveEscherichiacoli,three pairs of primers had been designed based on theO-antigengene inEnterohemorrhagicEscherichiacoliO157∶H7,ipaHgene inEnteroinvasiveEscherichiacolianduidAgene in allEscherichiacoli. The method was established and optimized,and the specificity and sensitivity were also analyzed. This method was applied to the detection of the artificially infected fresh lettuce with two pathogens. The results showed that the triplex PCR could detect two kinds of diarrheagenicEscherichiacoli. The sensitivity was 6.3×103CFU/mL. This method had high specificity and sensitivity. The sensitivity of the detection of artificial lettuce infected with two pathogens was 7.8×104CFU/mL. It was suggested that the method was suitable for rapid detection ofEnterohemorrhagicEscherichiacoliO157∶H7 andEnteroinvasiveEscherichiacoliin fresh agricultural products.
multiplex PCR;rapid detection;EnterohemorrhagicEscherichiacoliO157∶H7;EnteroinvasiveEscherichiacoli.
2015-01-28
董妍(1989-),女,在读硕士,研究方向:食品安全,E-mail:dong_yan@126.com。
*通讯作者:胡文忠(1959- ),男,博士,教授,研究方向:食品科学,E-mail:hwz@dlnu.edu.cn。
国家科技支撑计划项目(2012BAD38B05);国家自然科学基金项目(31172009);大连科技计划项目(2012E13SF106)。
TS201.3
A
1002-0306(2015)21-0312-07
10.13386/j.issn1002-0306.2015.21.056
猜你喜欢
中老年保健(2022年1期)2022-08-17 06:14:22
食品安全导刊(2021年20期)2021-08-30 06:39:04
中老年保健(2021年6期)2021-08-24 06:54:00
中国医疗保险(2017年5期)2017-05-17 08:26:39
现代食品(2016年24期)2016-04-28 08:11:54
系统工程与电子技术(2016年2期)2016-04-16 05:16:53
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
中国康复理论与实践(2015年10期)2015-12-24 05:42:46
现代电生理学杂志(2015年1期)2015-07-18 11:02:16
中国光学(2015年1期)2015-06-06 18:30:20
