
头孢拉定残留溶媒分析方法学的确认
2015-04-01 02:03董萍
机电信息 2015年5期
董 萍
(上海新亚药业有限公司新先锋药厂,上海201203)
0 引言
由于药品具有特殊性,因此对其进行检验方法学的确认是必须的,其可以确保样品分析的准确性、精密性,使广大民众可以放心安全地使用药品。根据新版GMP第223条规定,符合下列情形之一的,应当对检验方法进行验证:(1)采用新的检验方法;(2)检验方法需变更的;(3)采用《中华人民共和国药典》及其他法定标准未收载的检验方法;(4)法规规定的其他需要验证的检验方法。又根据《中华人民共和国药典(2010版增补本)》中残留溶媒测定项的要求,现对本公司生产的头孢拉定残留溶媒分析方法进行确认,以证明该分析方法在实验室条件下的适用性。
《中华人民共和国药典(2010版增补本)》规定对残留溶媒的测定可采用高效气相色谱法。高效气相色谱法(GC)以永久性气体为流动相(通常叫载气),利用流动相和固定相中的分配系数的差别,从而在两相间反复多次分配,使原来分配系数差别很小的各组分分离开来。气相色谱法的分析对象多为分子质量小于1 000、低沸点、易挥发、热稳定性好的化合物。
1 确认实验所用仪器与试剂/试药
1.1 实验所用的主要仪器
气相色谱仪:7890A,Agilent;顶空进样器:7697A,Agilent;分析天平:FA1604S,上海天平仪器厂;色谱柱:HP-1(60m×0.53mm×5 μm),Agilent;色谱工作站:Rev.C.01.03,Agilent。
1.2 实验所用的试剂/试药
丙酮(分析纯):500m L/瓶,宜兴市达华化工有限公司;头孢拉定(注射用):原料药,上海新亚药业有限公司。
2 确认的方法
2.1 色谱条件
检测温度:250℃;进样口温度:200℃;分流比:10:1;空气流速:400m L/min;氢气流速:40m L/min;氮气流速:15 psi(2.2 kPa);柱温:80℃;进样量:1.0 m L;顶空条件:顶空瓶10m L;平衡温度:80℃;平衡时间30m in;定量环温度:100℃;传输管温度:110℃。
2.2 供试品储备液配制
精密称取供试品1.0 g置于10m L容量瓶,加内标液至刻度,摇匀。
2.3 对照储备液配制
精密称取丙酮0.3 g置于50m L容量瓶,加水至刻度,摇匀。
2.4 供试品溶液配置
吸取供试品储备液3m L置于顶空瓶,密封瓶盖,摇匀。
2.5 对照品溶液配置
吸取对照品储备液10m L置于100m L容量瓶中,加水至刻度,再吸取3m L置于顶空瓶中,密封瓶盖,摇匀。
2.6 残留溶剂含量的计算
按外标法以峰面积计算残留溶剂含量:


3 确认内容
3.1 确认依据
《中华人民共和国药典(2010版增补本)》中关于残留溶媒测定项的要求。
3.2 专属性试验
吸取对照品储备液10m L置于100m L容量瓶中,加水至刻度,再吸取3m L置顶空瓶中,密封瓶盖,摇匀。制备对照品储备液5份,各进样1针,得到各物质峰面积的平均值的RSD(相对标准偏差)、分离度及保留时间。表1为各溶剂的峰面积、RSD、塔板理论数,图1~图5分别为各专属性色谱图,并根据下列公式计算相对标准偏差:

式中S——样本标准偏差;
x——计算结果的算术平均值。
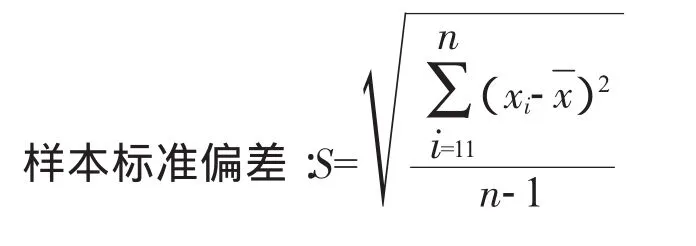
式中 n——样品总数;
i——物料中某成分的各次测量值。
根据表1数据,各溶剂峰的塔板理论数均大于5 000,符合验证要求。
3.3 定量限
以S/N=10计算定量限(其中,S为灵敏度,N为噪声),依次稀释对照品储备液。表2为丙酮定量限。
3.4 回收率
3.4.1 样品残留溶剂原有含量
1 g样品中残留溶剂的原有含量如表3所示。
3.4.2 重复性试验
精密移取储备液8m L置于100m L容量瓶,加水稀释至刻度,作为溶液1;精密移取储备液10m L置于100m L容量瓶,加水稀释至刻度,作为溶液2;精密移取储备液12m L置于100m L容量瓶,加水稀释至刻度,作为溶液3。分别取3份样品,每份1.0 g,分别置于10 m L容量瓶中,分别加入溶液1、溶液2、溶液3至刻度,作为样品溶液1、样品溶液2、样品溶液3,每种样品溶液吸取3m L置于顶空瓶,摇匀,各进3针。
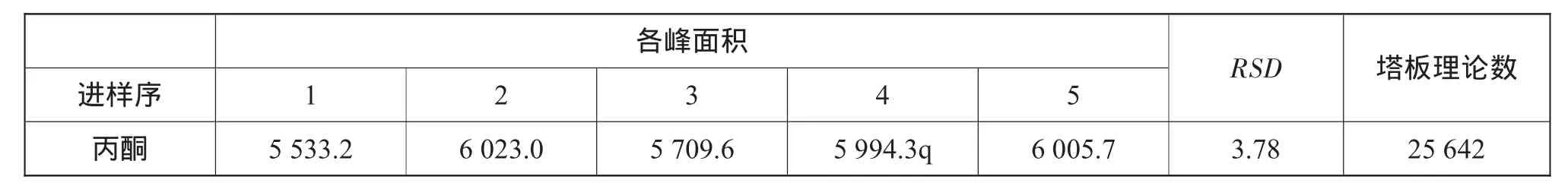
表1 各溶剂的峰面积、RSD、塔板理论数
图1 专属性色谱图1
图2 专属性色谱图2
图3 专属性色谱图3
图4 专属性色谱图4

图5 专属性色谱图5

表2 丙酮定量限
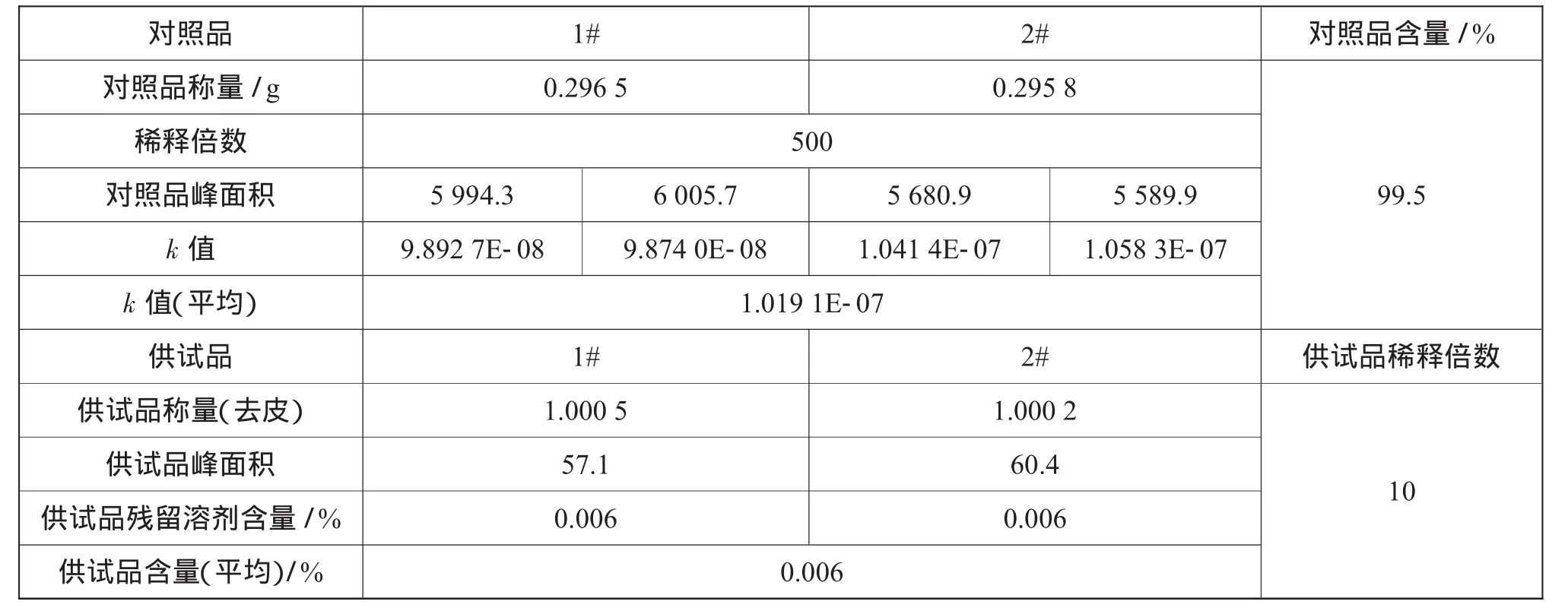
表3 1 g样品中残留溶剂原有含量
表4为根据称量得k平均值,图6~图14分别为回收率色谱图。
根据下列公式,分别计算各浓度溶液的回收率,并计算RSD值。表5为丙酮回收率。

表4 根据称量和峰面积得k平均值

图6 回收率色谱图1

图7 回收率色谱图2
图8 回收率色谱图3

图9 回收率色谱图4

图10 回收率色谱图5

图11 回收率色谱图6

图12 回收率色谱图7

图13 回收率色谱图8
图14 回收率色谱图9

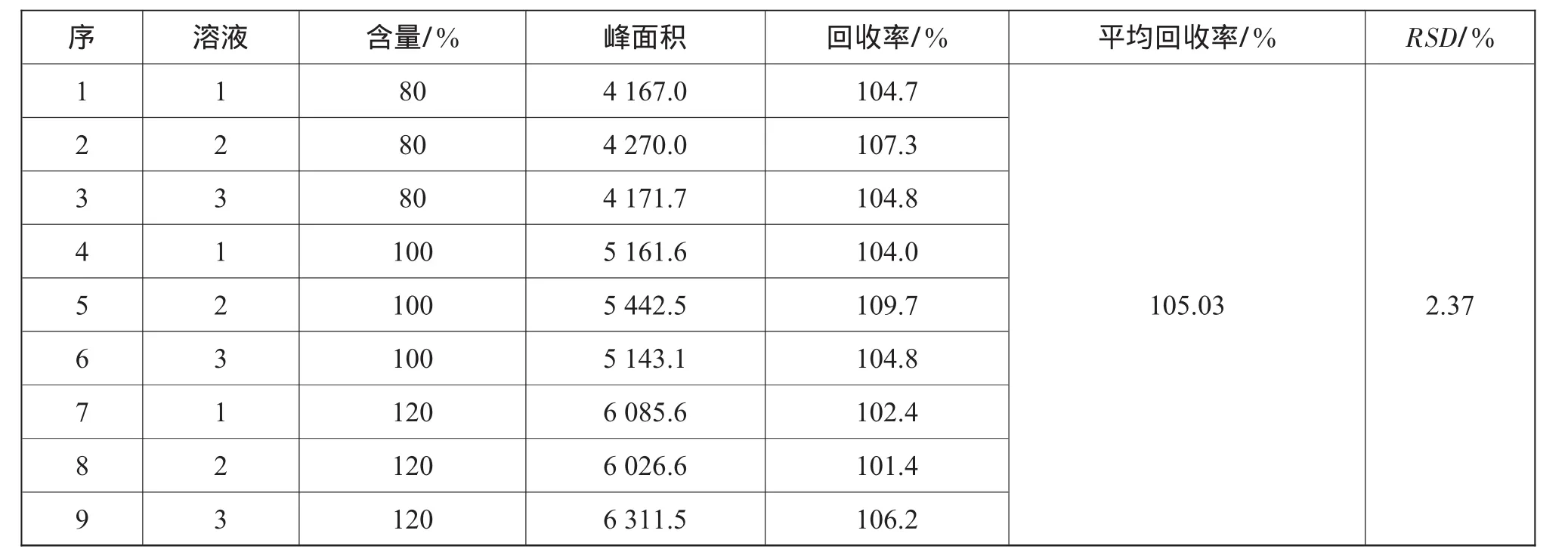
表5 丙酮回收率
3.5 精密度
连续进样6针,根据峰面积求RSD,表6为精密度。
4 结语
对本公司生产的头孢拉定残留溶媒分析方法学的确认结果汇总如表7所示。

表6 精密度
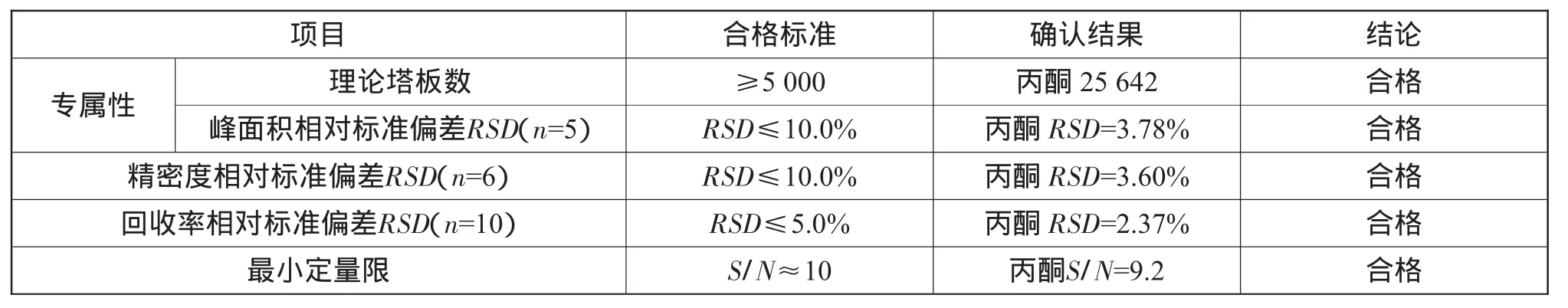
表7 确认结果汇总
从表7可知,以上方法学确认结果表明该检测方法准确、可靠,在本实验室条件下适用于头孢拉定残留溶媒的分析。
猜你喜欢
中国卫生标准管理(2022年13期)2022-07-29
临床合理用药杂志(2021年13期)2021-01-31
——一个解释欧姆表刻度不均匀的好方法
教学考试(高考物理)(2018年6期)2018-12-06
艺海(2018年2期)2018-08-29
科学与财富(2017年32期)2017-12-20
科学与财富(2017年29期)2017-12-20
数学小灵通(1-2年级)(2017年9期)2017-10-13
学苑创造·B版(2017年1期)2017-02-21
学术论坛(2016年5期)2016-05-17
小天使·二年级语数英综合(2016年9期)2016-05-14
