
双格氏试剂法合成4,4’-二(二苯基膦基)联苯
2015-03-27 09:41:58姚骏齐磊毕峰洪啸柳利李法宝
湖北大学学报(自然科学版) 2015年4期
姚骏,齐磊,毕峰,洪啸,柳利,李法宝
(有机化工新材料湖北省协同创新中心(湖北大学),有机功能分子合成与应用教育部重点实验室(湖北大学),湖北 武汉 430062)
0 引言
烷基膦中P的最外层电子具有3S23P3的电子构型,具有较强的电子给予能力,属于路易斯软碱[1],在配位化学中经常和Pd、Pt、Au、Ir和Cu等金属原子自组装形成具有各种结构的配位化合物[2-4].而磷原子上的孤对电子与芳香环的π共轭体系均可作为配位点,因此,双磷配体独特的分子结构使其配位性表现出良好的π-电子接受体特性,利用有机双膦配体来构建新颖结构的金属有机配合物也一直是化学工作者尤其是配位化学工作者研究的热点[5].有机双磷配体作为有机发光材料前体制备的金属配合物在电致发光OLED中有优异的表现.同时,以有机双膦化合物作为配体的金属配合物可作为各种重要反应的催化剂或催化剂前体,在催化领域也受到越来越多的关注[6].目前,双膦配体可通过光催化还原法[7],正丁基锂-卤素交换法[8]、钯催化剂催化法[9]等制备,这些方法虽然产率较高,但原材料价格昂贵,反应条件也较为苛刻[10].因此,探寻一种简便、温和、廉价的合成路线具有重要研究意义.
本文中通过设计合成双格氏试剂[11],并用于合成4,4’-二(二苯基膦基)联苯(图1).采用核磁共振谱(1H NMR、13C NMR和31P NMR)、傅里叶变换红外光谱(FT-IR)、液质联用飞行时间质谱(LC-MS TOF)、元素分析等对目标化合物进行了结构表征.
1 实验部分
1.1 仪器与试剂 仪器:美国瓦里安AS600(600 MHz)高分辨率核磁共振仪,中科开物公司WIPM-400(400 MHz)高分辨率核磁共振仪,Agilent 1260-6224 LC-MS TOF飞行时间液质联用仪、BX FI-IR型傅里叶转换红外光谱仪(美国Perkin-Elmet公司生产,KBr压片)、Vario Micro Cube元素分析仪、SHZ-Ⅲ型循环水式真空泵(上海荣生化学仪器厂)、恒温加热磁力搅拌器、AL-206型电子天平(上海,梅特勒-拖利多公司)、DZF-602型真空干燥箱(上海精宏实验设备有限公司)、磁力搅拌器、旋转蒸发仪、氮气保护装置.
试剂:THF经钠丝预干燥,加热回流至指示剂二苯甲酮呈蓝色后蒸出,镁条用稀盐酸浸泡除去表面氧化层,蒸馏水、乙醇洗涤后砂纸打磨剪碎,油泵抽干备用.所用试剂均为市售,试剂纯度均为分析.
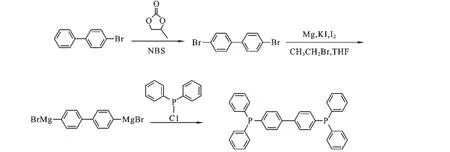
图1 4,4’-二(二苯基膦基)联苯的合成路线
1.2 4,4’-二溴联苯的合成 将4-溴联苯(10.0 g,43.1 mmol)加入100 mL碳酸丙烯酯中,60℃下搅拌,观察到固体全部溶解后,再向其中加入NBS(7.7 g,43.1 mmol),60℃下回流搅拌12 h后冷却到室温,有白色固体析出,过滤,蒸馏水洗涤干燥,得白色固体12.0 g,产率89.8%.1H NMR(400 MHz,CDCl3),δ=7.55(d,J=8.0 Hz,4H),7.42(d,J=8.0 Hz,4H).
1.3 4,4’-二(二苯基膦基)联苯的合成 氮气保护下,将Mg(3.70 g,154.2 mmol),KI(1.66 g,10.0 mmol)及一粒碘投入250 mL的三口瓶中,加入100 mL新蒸THF及2滴溴乙烷,微热至40℃引发反应,再缓慢滴入4,4’-二溴联苯(12.0 g,33.5 mmol)的THF溶液30 mL,升温至60℃反应12 h,降温至0℃,向反应液中滴加二苯基氯化磷(17.0 g,77.4 mmol),滴完后在室温搅拌4 h,加入50 mL氯化铵的饱和水溶液淬灭反应,过滤,用CH2Cl2萃取有机相,无水硫酸镁干燥后蒸干溶剂,柱层析分离(V二氯甲烷∶V石油醚=1∶5)得到白色固体5.02 g,该步骤产率为25.0%.IR(KBr,ν/cm-1):3 435,3 061,3 013,2 927,2 348,1 964,1 926,1 888,1 591,1 584,1 475,1 433,819,746,695.1H NMR(400 MHz,CDCl3),δ=7.56(d,J=8.0 Hz,4H),7.35~7.38 (m,24H),13C NMR(150 MHz,CDCl3),δ=140.92,137.30,136.71,134.45,134.31,134.04,133.90,129.00,128.77,128.73,127.28,127.23.31P NMR(240 MHz,CDCl3),δ=35.29.ESI-MS m/z calcd for C36H28P2[M+]522.166 6,found 522.115 4.C36H28P2元素分析计算值(%,实测值):C:82.74(82.76),H:5.40(5.35).
2 结果与讨论
2.1 IR谱 图2是目标产物的KBr压片的固体IR图谱.低频区中746和695 cm-1吸收带为一取代苯环C—H键弯曲振动所产生吸收峰,819 cm-1吸收带为对位二取代苯环C—H键弯曲振动所产生吸收峰,1 589,1 475和1 433 cm-1尖锐吸收带为芳环骨架振动峰;高频区3 061、3 013 cm-1为苯环 C—H的伸缩振动吸收峰.
2.21H NMR谱 图3为目标产物在CDCl3中的1H NMR图谱.联苯苯环上氢原子的化学位移在7.55~ 7.57之间出现共振信号,这是由于苯环的共轭效应及P的强给电子诱导效应共同作用,使二取代联苯苯环上远离P的4个氢原子共振吸收向低场移动.而7.38~7.35 ppm的多重峰为一取代苯及二取代联苯苯环上靠近P的苯环上氢原子的吸收峰,这是由于苯环H的化学环境相似导致的.每种氢对应的积分面积与结构相符.
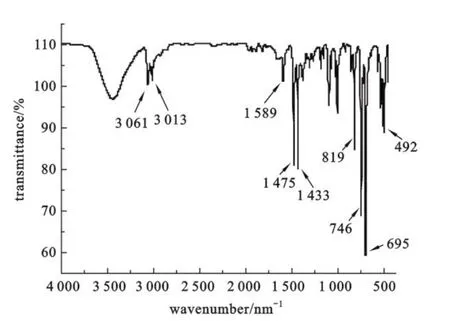
图2 4,4’-二(二苯基膦基)联苯的红外图谱

图3 4,4’-二(二苯基膦基)联苯的1H NMR图谱
2.313C NMR谱 图4为产物在CDCl3中的13C NMR图谱.苯环上碳原子化学位移在140.9~127.23之间发生共振信号,由于分子的对称性出现了8组峰,分别对应于苯环上的4个碳原子和联苯上的4个碳原子.由于P的给电子作用及苯环的共轭效应,联苯苯环上远离P的3个碳原子受到P的给电子作用影响较小,电子云密度较小,信号出现在低场,分别对应于140.92,137.30和136.71,而苯环上的4个碳原子和联苯上靠近P的3个碳原子受到P的给电子作用影响较大,电子云密度较大,信号出现在高场,对应于134.45~127.23.
2.431P NMR谱 图5为产物在CDCl3中的31P NMR图谱,化学位移为35.29处为苯环上的C—P单键产生的吸收峰,并且该吸收峰为一个单峰,说明2个P的化学环境相同,确证了该产物的分子对称性.与文献报道的芳基膦的C—P键的化学位移相符.[12]
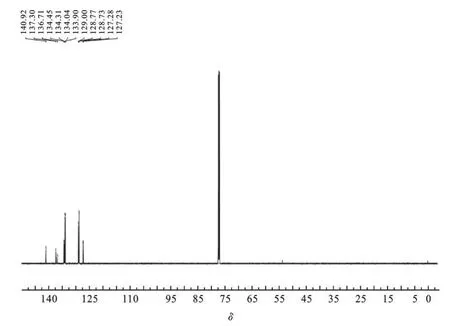
图4 4,4’-二(二苯基膦基)联苯的13C NMR图谱

图5 4,4’-二(二苯基膦基)联苯的31P NMR图谱
2.5 合成方法分析 本文中合成4,4’-二(二苯基膦基)联苯的步骤主要是制备双格氏试剂,无需分离直接与二苯基氯化磷反应,反应需在氮气氛下进行,反应温度为室温,反应时间4 h,整个反应过程及后处理比较简便,一步法合成了目标化合物.此外,整个反应采用的原料价格便宜、反应条件温和.采用柱层析技术分离容易,经柱层析纯化的目标化合物经核磁图、LC-MS-TOF检测,结构获得确证.
目前文献报道的芳基二膦化合物的合成方法,例如:光照法通过自由基反应上磷酸酯,再用LiAlH4等强还原剂还原[13],该方法对光照要求严格,对仪器装置要求较高,反应时间较长;采用正丁基锂进行卤素-锂交换法制备[14],该方法要求反应需在低温-66℃下进行,因为正丁基锂活性很高,常温下反应非常剧烈、不易控制;采用钯催化剂催化反应制备[15],但钯催化剂价格较为昂贵,大规模生产成本高.而本文中采用廉价的金属镁与二卤代物反应生成双格氏试剂,无需分离直接与二苯基氯化膦反应,反应条件温和,成本低,后处理也较为容易.
本文中所用合成方法也存在一些不足,如制备双格氏试剂时需要保证体系无水无氧,并注意防止产物被氧化而导致产率降低.
3 结论
本文中以光学活性的4-溴联苯为原料,通过卤代、制备双格氏试剂一步反应合成了目标化合物4,4’-二(二苯基膦基)联苯.采用核磁共振谱(1H NMR、13C NMR、31P NMR)、傅里叶变换红外光谱(FT-IR)、液质联用飞行时间质谱(LC-MS TOF)等对目标化合物进行了结构表征,确证了结构.与传统的合成法相比,本方法具有反应原料成本低、反应条件温和、操作简便等优点.该化合物作为具有良好发光性能的刚性有机配体,可与Pd、Pt、Au、Ir和Cu等金属合成有机膦金属配合物,在有机电致发光OLED及催化等领域有广泛的应用前景.
[1]Hashimoto M,Igawa S,Yashima M,et al.Highly efficient green organic light-emitting diodes containing luminescent threecoordinate copper(I)complexes[J].Journal of the American Chemical Society,2011,133(27):10348-10351.
[2]Huang Jin-kun,Chan J,Chen Ying,et al.A highly efficient palladium/copper cocatalytic system for direct arylation of heteroarenes:an unexpected effect of Cu(xantphos)I[J].Journal of the American Chemical Society,2010,132(11):3674.
[3]Lin Y Y,Wang Y J,Cheng Jun-hao,et al.Copper-catalyzed coupling of alkynes with alkenyl halides[J].Synlett,2012,23(6): 930-934.
[4]Kaufhold O,Stasch A,Edwards P G,et al.Template controlled synthesis of a coordinated[11]ane-P2CNHC macrocycle[J]. Chemical Communications,2007(18):1822-1824.
[5]Sapochak L,Burrows P.Organic materials with phosphine sulfide moieties having tunable electric and electroluminescent properties:U S Pat Appl Publ,20070241670[P].2007-10-18.
[6]Tunney S E,Stille A.Palladium-catalyzed coupling of aryl halides with(trimethylstannyl)diphenylphosphine and(trimethylsilyl) diphenylphosphine[J].Journal of Organic Chemistry,1987,52(5):748-753.
[7]Kyba Evan P.A facile synthesis of 1,2-Bis(phosphino)benzene and related alkylated species[J].Organometallics,1983(2): 1877-1879.
[8]Tanaka,Yousuke,Mashiko,et al.Novel diphosphonium salt,method for manufacturing the same,and antistatic agent and curing accelerator containing the same:PCT Int Appl,2014006828[P].2014-01-09.
[9]Davis P H,Belford R L,Paul L C.The crystal and molecular structure of bromobis(triphenylphosphine)copper(i)hemibenzenate[J].Inorganic Chemistty,1973,12(1):213-218.
[10]Yasukawa Keiich.Phosphines,and organic thin-film solar cells and tandem organic thin-film solar cells using them:Jpn Kokai Tokkyo Koho,2012028688[P].2012-02-09.
[11]Zhang Li,Liao Shi-jian,Xu Yun,et al.The reactivity of active magnesium from magnesium anthracene in the formation of digrignard reagents from aromatic dichlorids[J].Synthesis and Reactivity in Inorganic and Metal-Organic Chemistry,1996,26(5): 809-817.
[12]Sun Meng,Zhang Hongyu,Han Qi,et al.Nickel-catalyzed C-P cross-coupling by C-CN bond cleavage[J].Chemistry-A European Journal,2011,17(35):9566-9570.
[13]王作永.2,2'-Biphospholes:new building blocks for tuning the HOMO-LUMO gap[D].郑州:郑州大学,2013.
[14]许南南.基于磷杂苯骨架的双磷配体的设计、合成及表征[D].郑州:郑州大学,2012.
[15]Stol M,Snelders D J,Kooijman H,et al.A new,easily recyclable arylating agent based on a diphosphino-digold(I)complex[J].Dalton Transactions,2007(24):2589-2593.
猜你喜欢
中学化学(2022年5期)2022-06-17 16:51:48
高中数理化(2020年1期)2020-02-29 02:21:18
山东化工(2019年7期)2019-04-27 07:39:28
四川师范大学学报(自然科学版)(2018年2期)2018-04-28 02:21:08
中国资源综合利用(2017年1期)2018-01-22 02:44:25
中国塑料(2017年2期)2017-05-17 06:13:27
癌变·畸变·突变(2016年3期)2016-02-27 06:15:25
合成化学(2015年2期)2016-01-17 09:03:13
化工进展(2015年3期)2015-11-11 09:08:25
化学分析计量(2015年4期)2015-03-23 16:47:34
