
Structure and aromaticity of three-membered X3 (X=S, Se, Te) clusters
2015-03-23 08:08:23XIAOPengcheng,LIUYong,CHIXianxing
华中师范大学学报(自然科学版) 2015年1期
Structure and aromaticity of three-membered X3(X=S, Se, Te) clusters
The optimized geometries, vibration frequencies, atomization energies and total electronic energies of the three-membered X3(X=S, Se, Te) clusters are investigated at the B3LYP, MP2 and CCSD(T) levels of theory. To our knowledge, the theoretical study on these clusters is first reported here. Nucleus-independent chemical shifts (NICS) for these clusters indicate that a strong ring current exists in these three-membered structures (D3h). The detailed molecular orbital analysis for these isomers further reveals that one highly delocalized σ-type MOs and one π-type Mos which follow the 4n+2 electron counting rule respectively and play an important role in rendering these species two-fold aromaticity.
ab initio study; aromaticity; NICS
1 Introduction

In this article, we investigate theoretically a new class of neutral three-membered X3(X=S, Se, Te) clusters consisting of atoms S, Se and Te, for which there are two filled valence s-AO electrons and four valencep-AO electrons at the B3LYP, MP2 and CCSD(T) levels of theory. The aromaticity of these species by geometric structure analysis, NICS and MO analysis is explored. The research results show that a strong ring current exists in the three-membered rings of these species. One highly delocalized σ-type orbital and one π-type orbital in occupied molecular orbitals for these species respectively renders two-fold aromaticity, which adhere to the 4n+2 electron counting rule respectively. We believe in this work can provide some computational predictions for future possible experimental observation.
2 Computational methods
The structural optimization, vibration frequencies, and total electronic energies (Etot, including zero-point energies, ZPE) for the X3(X=S, Se, Te) clusters, are calculated with four theoretical methods: B3LYP, MP2 and CCSD(T). B3LYP is a DFT method using Beck’s three parameters functional (B3) along with the Lee, Yang, and Parr correlation functional (LYP). MP2 is a second-order Møller-Plesset perturbation method. CCSD(T) is the coupled cluster method using single, double, and triple substitutions from the Hartree-Fock determinant. Extended 6-311+G** basis set is used for S and Se atoms. LANL2DZ basis set including the effective core potential (ECP) and some relativistic effect is adopted for heavier metal atoms Te.
The magnetic characteristics are calculated with Nuclear Independent Chemical Shift (NICS) methods. NICS method is a magnetic criterion that mirrors the ring current. In this method, the nuclear magnetic resonance (NMR) parameters are calculated for a ghost atom, usually placed at the centre of ring, and the NICS value is the negative of the isotropic magnetic shielding constant at the ghost atom. System with negative NICS values are aromatic, since negative values arise when diatropic ring current (shielding) dominates, whereas systems with positive values are antiaromatic because positive value arise when paratropic current (deshielding) dominates. Non-aromatic cyclic system should, therefore, have NICS values around zero. The more negative the NICS, the more aromatic the system is. In this study four NICS values are calculated at four positions in each of all-metal clusters: the center of the ring and points 0.5 Å, 1.0 Å, 1.5 Å above the center of the ring, which are denoted NICS(0.0), NICS(0.5), NICS(1.0), NICS(1.5) respectively.
All calculations in this work are performed using the Gaussian03 program. The MO pictures are drawn using the Gaussiview 3.0 program.
3 Results and discussion
3.1 Optimized structures for X3(X=S, Se, Te) clusters
Two possible isomers (illustrated in Fig.1) with a regular trigonal structure (D3h) and linear structure (D∞h), respectively, for each of three clusters X3(X=S, Se, Te) are investigated with three methods: B3LYP/6-311+G*, MP2/6-311+G* and CCSD(T)/ 6-311+G*, except for Te3clusters, for which the basis set LANL2DZ is used, considering that Te is heavier metal atoms. The optimized geometrical parameters and total electronic energies (Etot, including ZPE) are summarized in Table 1, the calculated vibrational frequenciesviinTable2andthecalculatedNICSinTable3.
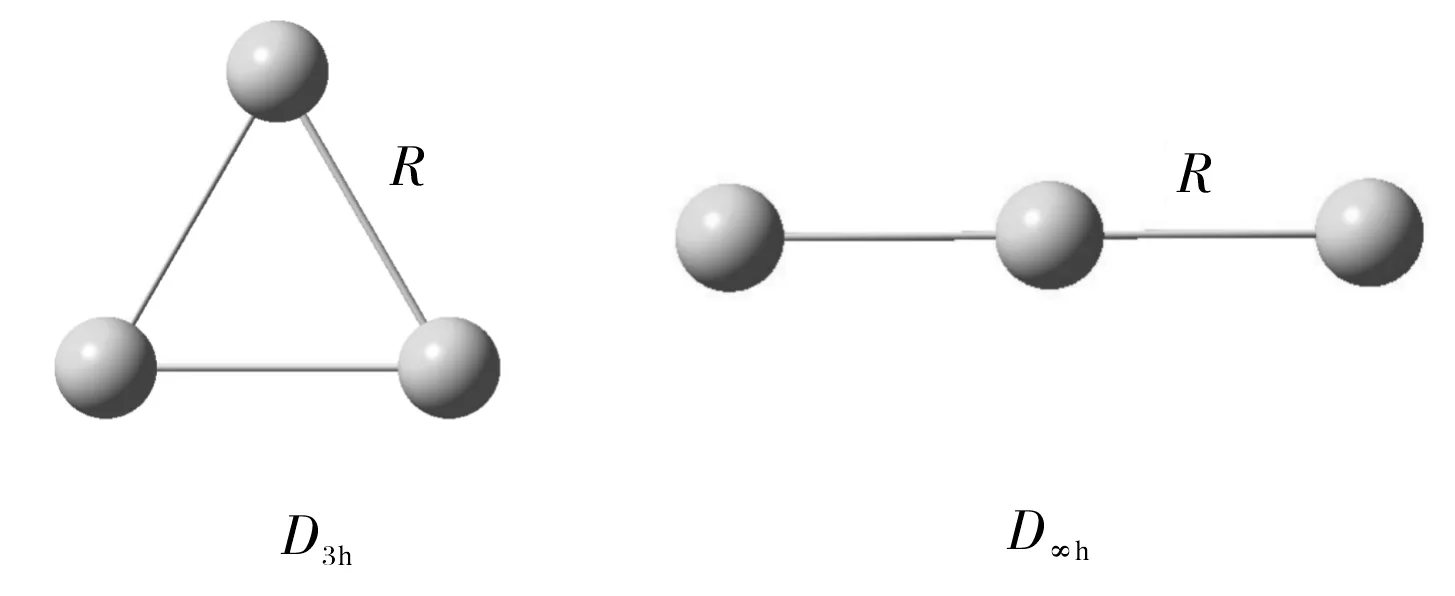
Fig.1 Optimized geometrical structures.
Thecalculatedresultsshowthatthereisnoimaginaryfrequencytoappearforthethreeregulartrigonalstructures(D3h), while there are two imaginary frequencies for each of the three linear structures (D∞h) with above three methods. So only the trigonal structures (D3h) are stable for the three X3(X=S, Se, Te) clusters. In addition, atomization energies (AE) of three X3(X=S, Se, Te) clusters are also calculated and the results are shown in table 1. TheAEvalues show that the sequence of these clusters arrranged in descending order ofAEsis S3>Se3>Te3for the trigonal structures (D3h) using above three method. The atomization energies (AEs) are unusually high, hinting at extra stability due to aromaticity. Note that theAEshave the big difference between MP2 method and other two methods(B3lyp and CCSD(T)). This is because MP2 method usually is used in the stronger interaction system.

Tab.1 The bond lengths R (Å), total electronic energies Etot (including ZPE,hartree), number of imaginary frequencies (Nimag), and atomization energy (AE in kcal/mol) for two structures: trigonal (D3h) and linear(D∞h) X3 (X=S, Se, Te) clusters.
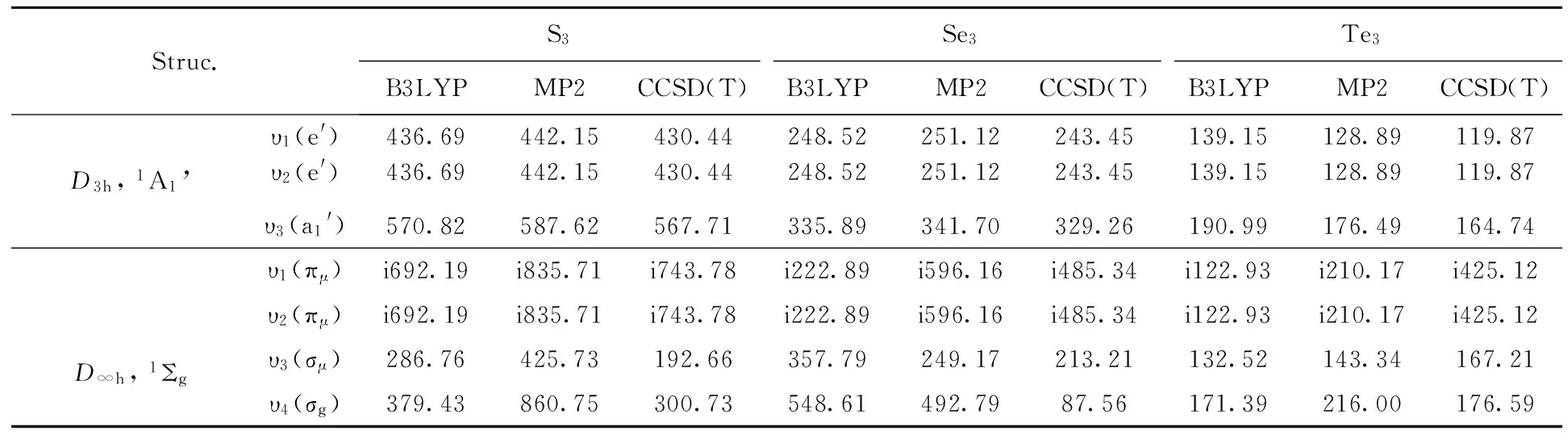
Tab.2 The calculated vibrational frequencies υi (cm-1) for two structures: trigonal (D3h) and linear (D∞h) X3(X=S, Se, Te) clusters. Lowercase letter i denotes imaginary frequencies.
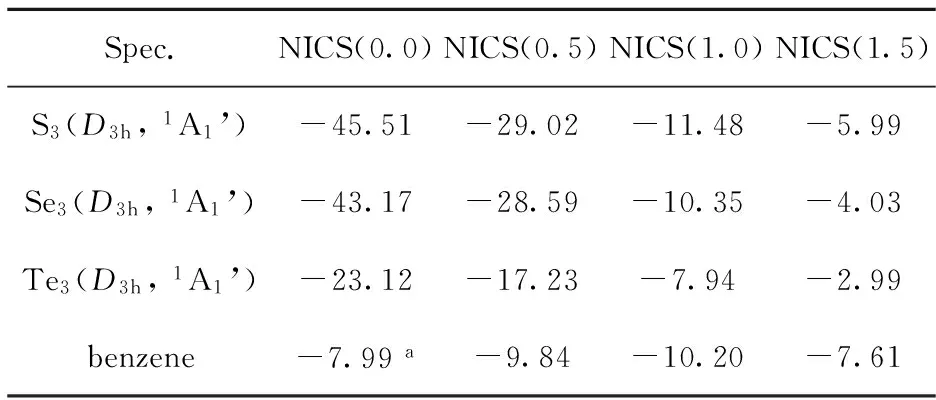
Tab.3 The calculated NICS (ppm cgsu) for trigonal(D3h) X3 (X=S, Se, Te) and benzene
3.2 Aromaticity of X3(X=S, Se, Te) clusters
3.2.1 Magnetic characteristics of aromaticity The calculations of NICS for X3(X=S, Se, Te) clusters are listed in Table 3. For comparison, the calculated NICS values for benzene are also listed in Table 3. For benzene, the NICS values are computed with B3LYP/6-311+G**//B3LYP/6-311+G**methods. The calculated NICS values show that the negative NICS values decrease in the order of NICS(0.0), NICS(0.5), NICS(1.0), and NICS(1.5) and are comparable to benzene. The NICS(0.0) values give an aromaticity ordering of S3>Se3>Te3, in the agreement with theAEordering of S3>Se3>Te3.
3.2.2 Molecular orbital analysis In this section, we will further explore the aromaticty of neutral X3(X=S, Se, Te) clusters by MO analysis. The valence electron configuration of X (X=S, Se, Te) atom is ns2np4. Therefore trigonal X3(X=S, Se, Te) clusters occupied nine valence MO orbitals: one HOMO and eight energetically lower valence MOs denoted HOMO-n(n=1-8) respectively. As shown in Fig.2, the occupied valence molecular orbitals of three clusters are very similar, just in the different sequences. Obviously, the HOMO-4 (a2") of Se3, Te3and HOMO-4 (a2") for S3is a π-bonding orbital formed from the out-of-plane 3pzorbitals. The HOMO-5 (a1′) for X3(X=S, Se, Te) is σ-bonding MO formed from the in-plane 3prorbitals. The following five MOs are the complete bonding, nonbonding, and antibonding ones made primarily by the same valence s AOs of corresponding X atoms. Their net bonding effect in expected to be zero and the s AOs can be viewed as lone pairs. Noteworthy is the presence of one highly delocalized π-type MOs (HOMO-4, a2") and one σ-type MOs(HOMO-5, a1′) for the three membered rings (Figure 2) which follow the 4n+2 π electron counting rule respectively and play an important role in rendering these species the two-fold aromaticity. On the basis of the above MO analysis, neutral X3(X=S, Se, Te) clusters all exhibit the multiple aromaticity.
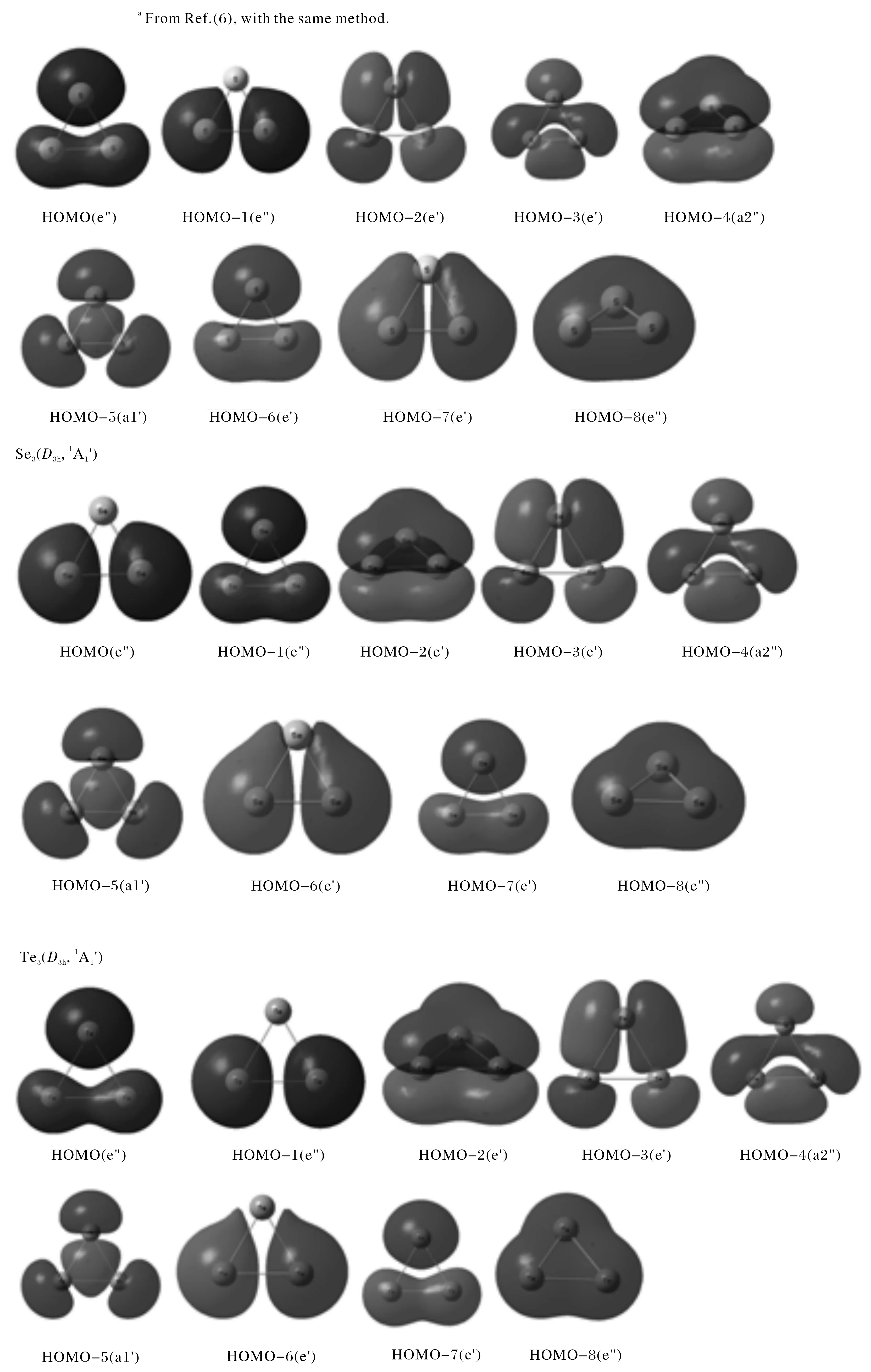
Fig.2 The occupied valence molecular orbital pictures for trigonal (D3h) X3 (X=S, Se, Te) clusters
4 Conclusion
In this paper, the equilibrium geometries, harmonic vibrational frequencies and electronic structure of the low-lying states of neutral X3(X=S, Se, Te) clusters are discussed. Comprehensive calculations show that neutral X3(X=S, Se, Te) clusters have only stable structure withD3hsymmetry respectively. The NICS values indicate that a strong diatropic ring current exists in the X3(X=S, Se, Te) clusters, suggesting they are highly aromatic. The detailed molecular orbital analyses confirm that X3(X=S, Se, Te) clusters have one delocalized π-MO and one delocalized σ-MOs. These findings are significant for expanding the aromaticity concept into inorganic X3(X=S, Se, Te) clusters.
[10] Schleyer P R, Jiao H. An evaluation of the aromaticity of inorganic rings: Refined evidence from magnetic properties [J]. J Am Chem Soc, 1997, 119: 12669-12670.
2014-08-19.
The Educational Commission of Hubei Province of China under Grant (B2014003).
1000-1190(2015)01-0087-05
XIAO Pengcheng1, LIU Yong1*, CHI Xianxing2
(1.Department of Mechanical and Electrical Engineering, Institute of Information Science and Technology,Hubei University of Education, Wuhan 430205;2.School of Physics and Electronic Information, Wenzhou University, Wenzhou,Zhejiang 325027)
三元团簇X3(X=S, Se, Te)的结构与芳香性
肖鹏程1, 刘 勇1, 池贤兴2
(1.湖北第二师范学院 物理与机械工程学院 信息科学与技术研究院, 武汉 430205;
2.温州大学 物理与电子信息学院, 浙江 温州 325027)
使用B3LYP, MP2 和 CCSD(T)3种方法研究了三元团簇 X3 (X=S, Se, Te)的几何结构,振动频率,原子化能和总能量.计算的核独立化学位移(NICS)指出在三元结构上存在强的电子环流.详细的分子轨道分析指出有一个高度离域化的σ和一个π分子轨道,它们在多重芳香性中有重要的作用.
从头算研究;芳香性;核独立化学位移
O562.1
A
*通讯联系人. E-mail: wjyliuyong@hue.edu.cn.
猜你喜欢
Acta Mathematica Scientia(English Series)(2022年5期)2022-11-04 09:06:40
Acta Mathematica Scientia(English Series)(2021年5期)2021-10-28 05:44:28
大学化学(2021年8期)2021-09-26 10:50:46
上海工艺美术(2021年4期)2021-04-24 21:30:13
Acta Mathematica Scientia(English Series)(2020年6期)2021-01-07 06:44:04
山东化工(2020年5期)2020-04-07 09:59:30
天津行政学院学报(2019年4期)2019-10-08 09:17:44
中外书摘(2017年2期)2017-02-10 19:42:15
湖南有色金属(2016年3期)2016-05-18 03:00:19
社会科学研究(2014年4期)2014-08-16 16:47:32
