
不同物相TiO2对H2O2/O3氧化效能的影响
2015-02-14 09:34倪金雷彭若帆童少平马淳安
化工学报 2015年10期
倪金雷,彭若帆,童少平,马淳安
(浙江工业大学化学工程学院,绿色化学合成技术国家重点实验室培育基地,浙江 杭州 310032)
不同物相TiO2对H2O2/O3氧化效能的影响
倪金雷,彭若帆,童少平,马淳安
(浙江工业大学化学工程学院,绿色化学合成技术国家重点实验室培育基地,浙江 杭州 310032)
研究了不同物相TiO2对H2O2/O3氧化效能的影响,目标有机物为羟基自由基探针化合物乙酸。结果表明,在初始pH为7.0和10.0时,加入TiO2反而降低了H2O2/O3的氧化效率,其中锐钛矿TiO2比金红石TiO2的减弱作用更为明显。当初始pH为3.0时,金红石TiO2能显著提高H2O2/O3的氧化效率,但锐钛矿TiO2影响不明显。机理分析表明,H2O2浓度及其衰减速率与乙酸的去除效率有很大的相关性。在pH为7.0和10.0时,两种物相TiO2均能加快H2O2的分解,其中锐钛矿TiO2作用更为显著。此条件下能有效引发臭氧分解产生羟基自由基,故H2O2过快分解反而降低了乙酸的去除效果。在pH为3.0时,H2O2去质子化反应困难,故O3/H2O2氧化效率极低,H2O2浓度也几乎不变。加入TiO2能明显提高H2O2的分解速率,相比金红石TiO2,锐钛矿TiO2使H2O2在5 min内基本分解完毕,但其对H2O2/O3氧化效率几乎没有影响。饱和臭氧水分解速度的批处理实验也有相似的结果。由此可见,合适引发剂浓度可能是保证臭氧类高级氧化技术较高效率的关键,否则只会导致氧化剂的无效过快分解。利用氯化硝基四氮唑蓝法对比分析了酸性条件下H2O2/O3、锐钛矿TiO2/H2O2/O3和金红石TiO2/H2O2/O3体系产生超氧自由基()的量,其大小顺序为:H2O2/O3< 金红石TiO2/H2O2/O3< 锐钛矿TiO2/H2O2/O3,这与前面结果吻合很好。
臭氧;双氧水;氧化;催化剂;二氧化钛;物相;自由基
引 言
臭氧氧化技术在水处理行业已经有一百多年的应用历史,臭氧氧化性的选择性又促进了臭氧类高级氧化技术的快速发展[1-2]。目前人们研究较多的臭氧类高级氧化技术包括H2O2/O3、UV/O3、超声/O3和催化臭氧化[3-13]。从应用的方便性和技术的成熟程度而言,H2O2/O3具有最好的应用前景[1,14-15]。然而,酸性条件下H2O2无法有效去质子化,故此条件下H2O2/O3的氧化效率极低[7,15]。事实上如何在酸性条件下有效产生羟基自由基是臭氧氧化技术的一个难点和重点[4]。在前期工作中发现加入Ti(Ⅳ)能有效提升H2O2/O3在酸性条件下的氧化效能[7-8],但该体系的不足是存在钛离子的流失问题。
将钛离子固定化处理是解决上述问题的一个关键。TiO2是一种稳定及无毒的半导体光催化剂,其在有机物的氧化降解中已有较多的研究[16-18]。利用TiO2催化臭氧化降解有机物也有一些报道[19-21]。仔细分析这些工作可以发现,所降解目标有机物往往有一定的臭氧化反应活性,一般在中碱性条件下有较好的结果[19,21]。另外,不同物相TiO2对该过程的影响问题也涉及较少[20]。然而已有结果表明,锐钛矿TiO2(anatase)的光催化氧化效能要明显优于金红石TiO2(rutile),它们与H2O2作用可在表面形成不同主体的活性氧物种[17]。尽管Fenton方法适合处理酸性废水,但其后续过程会有铁泥产生,pH适用范围也不宽。因此,对不同物相TiO2催化H2O2/O3氧化效能的研究有较好的实际意义,其中酸性条件下的结果对推动化工类废水的臭氧化处理意义更为突出。
乙酸(HAc)是化学氧化法降解有机物过程中的最终产物。在臭氧化反应过程中,考虑其反应特性,也常用作·OH的探针化合物[22]。因此,本文选用乙酸作为目标化合物进行降解,研究结果对广普性臭氧类高级氧化体系的建立具有重要意义。
1 实验材料与方法
1.1 材料与试剂
乙酸(CH3COOH)、双氧水(H2O2, 30%)、草酸钛钾(KTiO(C2O4)2·2H2O)、靛蓝胭脂红、氯化硝基四氮唑蓝(NBT)、磷酸(H3PO4)、硝酸(HNO3)、氢氧化钠(NaOH)和氢氧化钾(KOH)均为分析纯,购自华东医药。锐钛矿(anatase)TiO2和金红石(rutile)TiO2(粒径均为25 nm)购自于阿拉丁。实验中所有药品均购买后直接使用,所有溶液均用二次蒸馏水配制。
1.2 实验装置与过程
实验采用半批处理方式,装置如图1所示。管路、臭氧反应器和吸收器所用的材料为316L不锈钢、普通玻璃或聚四氟乙烯,连接部分采用硅胶管。臭氧发生器和破坏器的型号为CFS-1A和ODF-003(Ozonia,Switzerland),臭氧反应器(高为75 cm,内径为5 cm)为一带恒温夹套的玻璃反应器,布气装置为反应器底部的砂芯。
若无特殊说明,所有实验均在室温下进行,臭氧化氧气的流量为0.667 L·min−1,臭氧产量为41.4 mg·min−1,工作时水柱的高度约为30 cm。TiO2的加入量为4.0 g·L−1,其以悬浮状态存在。每次实验水样用容量瓶配制,实验体积一般为500 ml,对应HAc的初始浓度为200 mg·L−1,H2O2的初始浓度为100 mg·L−1(按100% H2O2计)。实验以通臭氧化氧气为计时起点。乙酸溶液的初始pH 用0.1 mol·L−1HNO3和0.1 mol·L−1NaOH调节。

图1 实验装置Fig.1 Diagram of experimental setup
1.3 分析方法
乙酸浓度采用离子色谱ICS-1500(戴安,美国)测定,色谱柱:AS19柱;淋洗液:1.008 g·L−1KOH,流速为1.0 ml·min−1。液相中H2O2浓度采用草酸钛钾法测定[23]。液相O3浓度用靛蓝胭脂红法测定[24]。pH 用pH精密酸度计测定。实验中定性对比了几个体系形成·O−2量的多少,·O−2用氯化硝基四氮唑蓝法测定[25]。其原理是现场产生的·O−2与氯化硝基四氮唑蓝(NBT)反应可以生成甲臜类化合物,在520 nm处有较宽的吸收峰,利用吸收峰的大小可以定量对比·O−2的产生量。具体实验过程:①配制150 mg·L−1H2O2和4 g·L−1TiO2于烧杯中,投加一定量的氯化硝基四氮唑蓝(0.05 mmol·L−1),搅拌2 h;②TiO2/H2O2体系中会有蓝色颗粒状物质生成,过滤,用一定量乙醇溶解,在紫外可见光下扫描甲臜的生成。
2 结果与讨论
2.1 初始pH为3.0时的氧化效率
初始pH为3.0时不同物相TiO2对H2O2/O3氧化乙酸效率的影响如图2所示。图2结果表明,金红石TiO2对臭氧及H2O2氧化乙酸的效率几乎没有影响,锐钛矿TiO2也有相似的结果。由于H2O2无法去质子化,故H2O2/O3对乙酸的降解效率也极低,30 min后只有5%左右。然而当溶液加入不同物相的TiO2时,乙酸降解效率有着明显的差异。锐钛矿TiO2对H2O2/O3氧化效率的影响几乎可以忽略;而金红石TiO2可以显著提高H2O2/O3的氧化效率,相同条件下30 min后乙酸的去除率达到32%左右。由于乙酸是臭氧化体系羟基自由基的探针化合物,故金红石TiO2有效促进了H2O2/O3体系·OH的形成。

图2 不同物相TiO2对乙酸降解效率的影响(pH=3.0)Fig.2 Effect of TiO2crystal phase on degradation efficiency of HAc (pH=3.0)
在此pH条件下TiO2/H2O2、H2O2/O3和TiO2/O3的氧化效率均极低,可以预计TiO2与H2O2的作用在TiO2/H2O2/O3中起了重要的作用。分析了实验过程H2O2浓度的变化情况,结果如图3所示。由于H2O2无法去质子化,故在实验时间范围内H2O2/O3体系H2O2浓度几乎保持不变;当溶液中加入金红石TiO2时,H2O2分解速度明显加快,30 min后分解率达50%左右,而锐钛矿TiO2可使H2O2在10 min内几乎分解完毕。基于氧化物中晶格钛对H2O2活化作用的研究结果[17,26-27],结合乙酸的降解结果及H2O2分解速度的变化情况,初步推测:在臭氧类高级氧化技术中,适当的引发剂浓度是保证有机物降解效率的关键(如本例中的金红石TiO2/H2O2/O3),过低(在此pH条件下如H2O2/O3)或过高的引发剂浓度(锐钛矿TiO2/H2O2/O3中)均不利于有机物降解效率的提高,过高的引发剂浓度会导致氧化剂的无效快速分解。
2.2 初始pH为7.0时的氧化效率
初始pH为7.0时不同物相TiO2对H2O2/O3氧化效率的影响如图4所示。在初始pH为7.0时,H2O2可以实现去质子化反应形成共轭碱,而是引发臭氧分解产生·OH的良好引发剂[15,28-29]。因此,在此条件下H2O2/O3能够快速降解水中的乙酸。然而当溶液中加入不同物相的TiO2后,无论是金红石TiO2还是锐钛矿TiO2,乙酸的降解效率反而有了明显的下降。相比金红石TiO2,锐钛矿TiO2的减弱作用更为显著。推测加入TiO2可能导致了溶液中H2O2的过快无效分解,进而导致乙酸降解效率的下降。
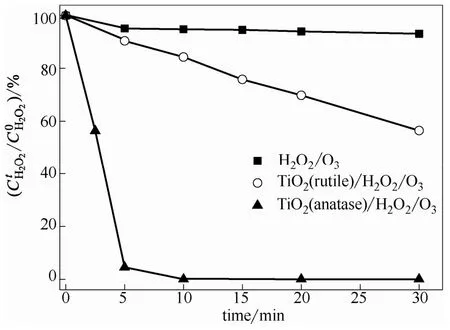
图3 乙酸降解过程H2O2浓度的变化情况(pH=3.0)Fig.3 Evolution of H2O2concentration in process of HAcdegradation (pH=3.0)

图4 不同物相TiO2对乙酸降解效率的影响(pH=7.0)Fig.4 Effect of TiO2crystal phase on degradation efficiency of HAc (pH=7.0)
为了证实上述推测,分析了此pH条件下H2O2浓度的变化情况,结果如图5所示。由图可知,TiO2的加入确实加快了水中H2O2的分解速度,其中锐钛矿TiO2的促进作用更明显。如H2O2在H2O2/O3、金红石TiO2/H2O2/O3和锐钛矿TiO2/H2O2/O3体系中分解完毕所需的时间分别约为30、20和10 min,这个结果与乙酸的降解效率有着很好的相关性。当H2O2分解完毕后,溶液中自由基引发剂(包括及钛-H2O2络合氧物种)失去了来源,此时羟基自由基也无法形成,从而导致实验后期乙酸的浓度几乎不变(乙酸与臭氧的反应速率常数<3×10−5L·mol−1·s−1)。

图5 乙酸降解过程H2O2浓度的变化情况(pH=7.0)Fig.5 Evolution of H2O2concentration in process of HAc degradation (pH=7.0)
2.3 初始pH为10.0时的氧化效率
在初始pH为10.0条件下,对比了几个氧化体系乙酸的降解效率和H2O2的分解速度,结果如图6和图7所示,其与pH=7.0时有相似的结果。另外,对比两个pH条件下乙酸的最终去除率和H2O2的分解情况,发现pH为7.0时H2O2/O3具有更高的降解效率,H2O2的分解速度也相对慢一点。该结果也支持了前文的推测,即合适的引发剂浓度是臭氧类高级氧化技术降解效率提高的关键,否则会导致氧化剂的无用消耗。
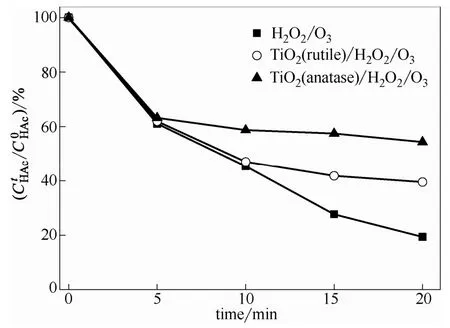
图6 不同物相TiO2对乙酸降解效率的影响(pH=10.0)Fig.6 Effect of TiO2crystal phase on degradation efficiency of HAc (pH=10.0)
2.4 初始pH为3.0时TiO2/H2O2/O3的作用机制
由以上结果可知,中碱性溶液加入TiO2会使H2O2过快分解,从而导致乙酸降解效率的下降。在酸性溶液中,尽管加入锐钛矿TiO2仍然存在H2O2分解过快的问题,但金红石TiO2使H2O2有一个较合适的分解速度,H2O2/O3氧化效率也提升明显。以上结果对酸性难降解废水的有效(预)处理意义十分突出。从3个pH条件的实验结果可以看出,不同物相TiO2在不同pH条件下都能加快H2O2的分解速度。有关氧化物晶格中钛与H2O2的作用问题已经有较多的研究报道,一般认为体系会产生一些活性氧物种[17,26-27]。根据水中臭氧的分解机理[30],这类活性氧物种往往能有效引发臭氧分解产生羟基自由基。

图7 乙酸降解过程H2O2浓度的变化情况(pH=10.0)Fig.7 Evolution of H2O2concentration in process of HAc degradation (pH=10.0)
根据以上分析,工作中利用氯化硝基四氮唑蓝法对比了3个体系超氧化自由基离子的产生量。该方法的原理是氯化硝基四氮唑蓝会与反应形成蓝色不溶于水的甲臜,过滤后利用有机溶剂溶解可以在520 nm附近产生明显的吸收峰,该峰值大小可用于定量对比的产生量。图8为H2O2、金红石TiO2/H2O2和锐钛矿TiO2/H2O23种溶液中在2 h产生量的对比,其大小顺序为:锐钛矿TiO2/H2O2> 金红石TiO2/H2O2> H2O2。该pH条件下单纯H2O2/O3体系H2O2和乙酸浓度几乎不变,因此TiO2与H2O2作用形成的活性氧物种不但是H2O2过快分解的主要原因,也是酸性条件H2O2/O3能有效降解乙酸的主要原因。上述活性氧物种产生量大小的顺序也与前面乙酸降解及H2O2分解的结果相一致。

图8 3种溶液甲臜产生量的对比Fig.8 Comparison in amount of formazan for three solutions
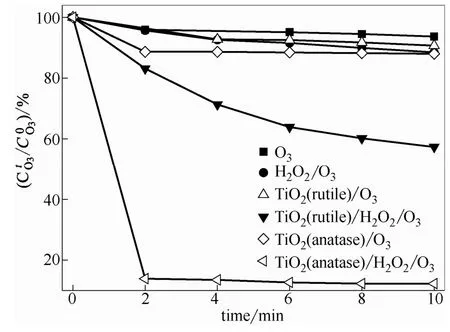
图9 批处理实验不同条件下水中臭氧浓度的变化情况Fig. 9 Evolution of ozone concentration in water under different conditions in batch test
由于超氧化物能有效地触发臭氧分解产生羟基自由基,因此预计不同体系水中的臭氧浓度也会有明显不同的结果。工作中分别在批处理(将一定体积比的饱和臭氧水与特定条件的乙酸溶液混合,在磁力搅拌条件下测定水中臭氧浓度的变化)和半批处理(即液相静态,臭氧化氧气连续)方式下分别对比了不同氧化体系水中臭氧浓度的变化情况。批处理实验结果如图9所示,单独加入H2O2或不同物相TiO2时,水中O3分解速度极为缓慢;当溶液中同时存在TiO2和H2O2时,O3分解速率明显加快,其中锐钛矿TiO2可使溶液中的臭氧在2 min内几乎分解完毕。半批处理实验结果如图10所示,由于TiO2/H2O2/O3体系存在引发臭氧分解的活性氧物种,故在实验时间范围内均低于单独臭氧过程水中的臭氧浓度。另外,由于锐钛矿TiO2使H2O2过快分解,故一定时间后溶液臭氧浓度很快上升。由此可见,锐钛矿TiO2/H2O2/O3中引发剂的浓度过高,反而使H2O2的分解速度过快,进而导致体系氧化效能的降低。

图10 半批处理实验中水中臭氧浓度的变化Fig. 10 Evolution of ozone concentration in water in semi-batch test
3 结 论
(1)在初始pH为7.0和10.0条件下,加入不同物相的TiO2反而降低了H2O2/O3氧化乙酸的效率,其中锐钛矿TiO2的减弱作用更为明显,其原因可能是加入TiO2使溶液中的H2O2发生快速分解,体系无法形成分解臭氧的自由基引发剂。
(2)在初始pH为3.0时,H2O2/O3对乙酸几乎无降解,实验过程H2O2浓度也几乎不变。加入金红石TiO2可使H2O2/O3氧化乙酸的效率明显提高,但加入锐钛矿TiO2对H2O2/O3的氧化效率几乎没有影响。与加入金红石TiO2相比,加入锐钛矿TiO2可使H2O2在短时间内分解完毕,使得溶液无法形成自由基引发剂,直接导致乙酸降解效率的降低。该结果有助于酸性条件下高效臭氧类高级氧化体系的创建。
(3)超氧自由基离子的分析结果表明,在超氧自由基离子的产生量上,不同物相TiO2有不同结果,其结果如下:锐钛矿TiO2/H2O2> 金红石TiO2/H2O2> H2O2。无论是饱和臭氧水的臭氧衰减速率情况,还是半批处理实验水中臭氧浓度的变化情况,均与该结果相一致。
[1] Legube B, Leitner N K V. Catalytic ozonation: a promising advanced oxidation technology for water treatment [J].Catalysis Today, 1999, 53(1): 61-72.
[2] Kasprzyk-Hordern B, Ziólek M, Nawrocki J. Catalytic ozonation and methods of enhancing molecular ozone reactions in water treatment [J].Applied Catalysis B:Environmental, 2003, 46(4): 639-669.
[3] Zeng Z Q, Wang J F, Li Z H, Sun B C, Shao L, Li W J, Chen J F, Zou H K. The advanced oxidation process of phenol solution by O3/H2O2in a rotating packed bed [J].Ozone:Science & Engineering, 2013, 35(2): 101-108.
[4] Nawrocki J. Catalytic ozonation in water: controversies and questions. discussion paper [J].Applied Catalysis B:Environmental, 2013, 142-143: 465-471.
[5] Pines D S, Reckhow D A. Effect of dissolved cobalt(Ⅱ) on the ozonation of oxalic acid [J].Environmental Science and Technology, 2002, 36(19): 4046-4051.
[6] Beltrán F J, Rivas F J, Montero-de-Espinosa R. Iron type catalysts for the ozonation of oxalic acid in water [J].Water Research, 2005, 39(15): 3553-3564.
[7] Li Wenwen(李文文), Liu Pengpeng(刘朋朋), Zhang Hua(张华), Shi Rui(石锐), Tong Shaoping(童少平), Ma Chunʼan(马淳安). Degradation of acetic acid by Ti(Ⅳ)-catalyzed H2O2/O3[J].CIESC Journal(化工学报), 2010, 61(7): 1790-1795.
[8] Tong S P, Zhao S Q, Lan X F, Ma C A. A kinetic model of Ti(Ⅳ)-catalyzed H2O2/O3process in aqueous solution [J].Journal of Environmental Sciences, 2011, 23(12): 2087-2092.
[9] Xiao H, Liu R P, Zhao X, Qu J H. Enhanced degradation of 2,4-dinitrotoluene by ozonation in the presence of manganese(Ⅱ) and oxalic acid [J].Journal of Molecular Catalysis A:Chemical, 2008, 286(1/2): 149-155.
[10] Dorota B M, Jacek S, Stanislaw L. Kinetic studies ofn-butylparaben degradation in H2O2/UV system [J].Ozone:Science & Engineering, 2012, 34(5): 354-358.
[11] Qi F, Xu B B, Chen Z L, Ma J, Sun D Z, Zhang L Q. Influence of aluminum oxides surface properties on catalyzed ozonation of 2,4,6-trichloroanisole [J].Separation and Purification Technology, 2009, 66: 405-410.
[12] Zhang T, Li C J, Ma J, Tian H, Qiang Z M. Surface hydroxyl groups of syntheticα-FeOOH in promoting ·OH generation from aqueous ozone: property and activity relationship [J].Applied Catalysis B:Environmental, 2008, 82: 131-137.
[13] Ikhlaq A, Brown D R, Kasprzyk-Hordern B. Mechanisms of catalytic ozonation: an investigation into superoxide ion radical and hydrogen peroxide formation during catalytic ozonation on alumina and zeolites in water [J].Applied Catalysis B:Environmental, 2013, 129: 437-449.
[14] Bauman M, Lobnik A, Hribernik A. Decolorization and modeling of synthetic wastewater using O3and H2O2/O3processes [J].Ozone:Science & Engineering, 2011, 33(1): 23-30.
[15] Kurniawan T A, Lo W H, Chan G Y S. Radicals-catalyzed oxidation reactions for degradation of recalcitrant compounds from landfill leachate [J].Chemical Engineering Journal, 2006, 125(1): 35-57.
[16] Inagaki M, Nonaka R, Tryba B, Morawski A W. Dependence of photocatalytic activity of anatase powders on their crystallinity [J].Chemosphere, 2006, 64: 437-445.
[17] Hirakawa T, Yawata K, Nosaka Y. Photocatalytic reactivity for·and·OH radical formation in anatase and rutile TiO2suspension as the effect of H2O2addition [J].Applied Catalysis A:General, 2007, 325: 105-111.
[18] Daimon T, Hirakawa T, Kitazawa M, Suetake J, Nosaka Y. Formation of singlet molecular oxygen associated with the formation of superoxide radicals in aqueous suspensions of TiO2photocatalysts [J].Applied Catalysis A:General, 2008, 340: 169-175.
[19] Song S, Liu Z W, He Z Q, Zhang A L, Chen J M, Yang Y P, Xu X H. Impacts of morphology and crystallite phases of titanium oxide on the catalytic ozonation of phenol [J].Environmental Science and Technology, 2010, 44: 3913-3918.
[20] Rosal R, Rodríguez A, Gonzalo M S, García-Calvo E. Catalytic ozonation of naproxen and carbamazepine on titanium dioxide [J].Applied Catalysis B:Environmental, 2008, 84: 48-57.
[21] Yang Y, Ma J, Qin Q, Zhai X. Degradation of nitrobenzene by nano-TiO2catalyzed ozonation [J].Journal of Molecular Catalysis A:Chemical, 2007, 267: 41-48.
[22] Jerry C, Meryer P A, Marrone J W T. Acetic acid oxidation and hydrolysis supercritical water [J].AIChE Journal, 1995, 41(9): 2108-2121.
[23] Sellers R M. Spectrophotometric determination of hydrogen peroxide using potassium titanium (Ⅳ) oxalate [J].The Analyst, 1980, 150(1255): 950-954.
[24] Bader H, Hoigne J. Determination of ozone in water by the indigo method [J].Water Research, 1981, 15(4): 449-456.
[25] Merchant M, Hardy R, Williams S. Quantitative detection of superoxide ions in whole blood of the American alligator (alligator mississippiensis) [J].Spectroscopy Letters, 2008, 41: 199-203.
[26] Hulea V, Dumitriu E, Patcas F, Ropot R, Graffin Patrick, Moreau Patrice. Cyclopentene oxidation with H2O2over Ti-containing zeolites [J].Applied Catalysis A:General, 1998, 170: 169-175.
[27] Casuscelli S G, Eimer G A, Canepa A, Heredia A C, Poncio C E, Crivello M E, Perez C F, Aguilar A, Herrero E R. Ti-MCM-41 as catalyst for a-pinene oxidation study of the effect of Ti content and H2O2addition on activity and selectivity [J].Catalysis Today, 2008, 133/134/135: 678-683.
[28] Lanao M, Ormad M P, Ibarz C, Miguel N, Ovelleiro J L. Bactericidal effectiveness of O3, O3/H2O2and O3/TiO2on clostridium perfringens [J].Ozone:Science & Engineering, 2008, 30(6): 431-438.
[29] Moussavi G, Yazdanbakhsh A, Heidarizad M. The removal of formaldehyde from concentrated synthetic wastewater using O3/MgO/H2O2process integrated with the biological treatment [J].Journal of Hazardous Materials, 2009, 171(1/2/3): 907-913.
[30] Staehelin J, Hoigne J. Decomposition of ozone in water in the presence of organic solutes acting as promoters and inhibitors of radical chain reactions [J].Environmental Science and Technology, 1985, 19(12): 1206-1213.
Effect of TiO2crystal phase on oxidation efficiency of H2O2/O3
NI Jinlei, PENG Ruofan, TONG Shaoping, MA Chun'an
(State Key Laboratory Breeding Base of Green Chemistry-Synthesis Technology,College of Chemical Engineering,Zhejiang University of Technology,Hangzhou310032,Zhejiang,China)
The oxidation efficiency of H2O2/O3catalyzed by titanium dioxide (TiO2) for acetic acid (HAc) degradation, a probe compound for hydroxyl radical in ozonation, was investigated, with a focus on the effect of TiO2crystal phase. The results indicated that the addition of TiO2showed negative effect on the oxidation efficiency when the initial pH was at 7.0 and 10.0, and among all crystal phases of TiO2anatase had the biggest negative effect. However, when the initial pH was at 3.0, rutile could significantly improve the oxidation efficiency of the H2O2/O3system, and anatase had negligible effect. The mechanism study showed that there existed a good correlation between degradation rate of HAc and concentration of H2O2(or its decomposition rate). Both anatase and rutile could accelerate decomposition of H2O2at initial pH of 7.0 and 10.0, and faster was for the former than the latter. Too high decomposition rate of H2O2could reduce removal rate of HAc at the two pH,because the conjugate baseof H2O2generated could react with ozone to effectively produce hydroxyl radicals (·OH). At initial pH 3.0, the oxidation efficiency of H2O2/O3system was very low due to the difficulty of H2O2deprotonation, so the concentration of H2O2had almost no change. Addition of TiO2could markedly accelerate the decomposition rate of H2O2, including deprotonation step, and anatase made H2O2decomposition finish in 5 min and too fast, leading to have no effect on the oxidation efficiency. However, rutile had no such high decomposition rate for H2O2and could generate-similar species which could react with ozone to produce hydroxyl radicals (·OH) to degrade acetic acid. The batch test carried out also gave a similar result. Therefore, it can be concluded that suitable initiator and its concentration may play an important role in ozone-based advanced oxidation process, and that too high concentration of initiator might lead to rapid consumption of oxidants. The amounts of superoxide ion radical () in H2O2/O3, anatase TiO2/H2O2/O3and rutile TiO2/H2O2/O3systems were determined by capturing method of Nitro Blue Tetrazolium Chloride (NBT), the order was as follows: H2O2/O3<rutile TiO2/H2O2/O3< anatase TiO2/H2O2/O3, which was in accord with the results of the results of HAc degradation.
ozone; hydrogen peroxide; oxidation; catalyst; titanium dioxide; crystal phase; radical
Prof. TONG Shaoping, sptong@zjut.edu.cn
10.11949/j.issn.0438-1157.20141858
X 703
:A
:0438—1157(2015)10—3950—07
2014-12-16收到初稿,2015-05-04收到修改稿。
联系人:童少平。
:倪金雷(1989—),男,硕士研究生。
国家自然科学基金项目(21176225);浙江省重点科技专项(2013C03019)。
Received date: 2014-12-16.
Foundation item: supported by the National Natural Science Foundation of China (21176225) and the Special Major Project of Science and Technology of Zhejiang Province(2013C03019).
猜你喜欢
四川地质学报(2022年2期)2022-07-08
农业研究与应用(2021年2期)2021-08-12
矿产勘查(2020年8期)2020-12-25
世界有色金属(2020年4期)2020-05-16
原子与分子物理学报(2020年5期)2020-03-17
——以金红石为例
中国金属通报(2020年23期)2020-03-15
无机盐工业(2017年6期)2017-03-11
无机盐工业(2016年6期)2016-03-15
无机盐工业(2016年2期)2016-03-15
湖南大学学报·自然科学版(2014年7期)2014-11-28
