
基于密度泛函的硫黄S8开环裂解机理
2015-02-14 09:34王荣杰沈本贤马健赵基钢
化工学报 2015年10期
王荣杰,沈本贤,马健,赵基钢
(1华东理工大学化学工程联合国家重点实验室,上海 200237;2石河子大学化学化工学院,新疆生产建设兵团化工绿色过程重点实验室,新疆 石河子 832003)
基于密度泛函的硫黄S8开环裂解机理
王荣杰1,2,沈本贤1,马健1,赵基钢1
(1华东理工大学化学工程联合国家重点实验室,上海 200237;2石河子大学化学化工学院,新疆生产建设兵团化工绿色过程重点实验室,新疆 石河子 832003)
采用基于密度泛函理论的量子化学方法研究了气化法制备不溶性硫黄过程中硫黄(S8)的开环裂解机理,建立了可能的反应路径,通过过渡态理论,计算了各个基元反应的活化能与反应速率常数,并对各个基元反应进行了动力学模拟计算,得到气体硫黄产物分布。研究结果表明,硫黄S8优先裂解成·S2·和·S6·,反应能垒为209.45 kJ·mol−1,气态硫黄S8开环裂解后生成产物主要为·S2·、·S4·和·S6·,与蒸汽密度法得到的组成分布相似,为进一步研究不溶性硫黄分子结构及聚合机理提供了参考方向。
不溶性硫黄;开环裂解;反应机理;密度泛函;数值模拟
引 言
近年来,随着子午线轮胎的快速发展,在钢丝帘布胶中以不喷出为特点的不溶性硫黄得到大量应用[1],不溶性硫黄除了可解决在轮胎橡胶表面的喷出问题之外,在非轮胎橡胶制品方面也克服了普通硫黄分散性不均的缺点,大量应用于胶鞋、橡胶地板、胶布中,现已占据橡胶硫化剂的大部分市场。
不溶性硫黄又称聚合硫(insoluble sulfur, IS),是硫的均聚物。不溶性硫黄是指不溶于二硫化碳的聚合硫黄,是普通硫黄的一种高分子改性品种,分为充油型和未充油型两大类,目前工业化制备方法主要有气化法和熔融法[2],前人进行了大量的实验研究以提高不溶性硫黄的产率和稳定性[3],但关于气化法不溶性硫黄制备机理特别是溶质硫黄开环裂解机理的研究较少,Jones等[4]利用密度泛函和Monte Carlo方法研究了Sn(n=2~18)的结构,袁洪娟[5]根据实验结果提出了开环裂解机理,关于硫黄开环聚合的热力学计算也有了一定的进展[6]。但由于实验条件的限制,利用实验方法尚不能得到硫黄裂解的详细反应历程,在分子尺度水平模拟方面,尚未见采用理论方法对硫黄开环裂解反应进行深入研究。近几年来,分子模拟技术在许多领域中得到了广泛的应用[7-11]。采用分子模拟技术研究硫黄分子的热裂解反应机理,可更深层次地理解其化学反应过程,确定分子结构及预测化学反应体系的特性。为此,采用密度泛函方法结合数值编程计算方法对硫黄开环裂解反应机理进行了深入研究,通过模拟硫黄开环断裂过程机理,为进一步探索不溶性硫黄生成机理及其结构奠定了基础。
1 模拟及计算方法
采用Materials Studio 7.0模拟软件的Dmol3模块,该模块以密度泛函理论为基础,参照前人[12-14]在热裂解体系分子模拟方面的研究经验,计算方法选择GGA,泛函形式选择PW91,基组为DND,由于模拟计算过程涉及到自由基,故所有电子自旋选用Spin unrestricted,自旋多重度选择Auto。
模拟步骤如下:首先对所有的反应物和生成物进行基于密度泛函理论的几何优化,采用LST/QST搜索各基元反应过渡态,分析频率确定只有一个虚频,说明过渡态搜索正确。
模拟计算各基元反应的反应能垒,探索能垒最低的最优反应路径。
根据相关文献,气态硫黄的分子组成主要有·S2·、·S4·、·S6·、S8,在生产不溶性硫黄时,反应温度为400~800℃,故1500℃以上才能裂解出现的单原子硫不予考虑。假设所有的裂解反应均只生成两种产物,故可能存在的反应有如下9种。

采用分子模拟方法对上述9种气态硫黄裂解过程中可能存在的反应进行了分子模拟,对于基元反应,根据化学反应过渡态理论[15]可知活化焓、活化能、反应速率k分别为
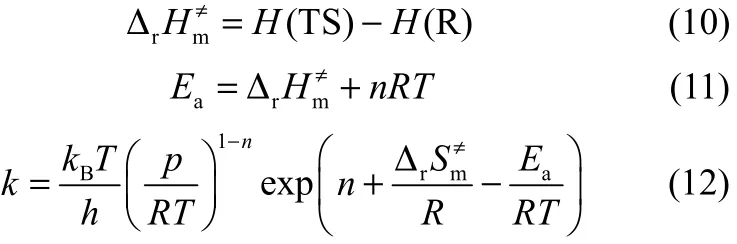
由过渡态理论可计算上述基元反应活化能以及在某一温度下的反应速率常数,并根据各组分的物料平衡和动力学方程导出反应模型动力学方程组,采用Runge-Kutta法编程进行数值求解。
2 结果与讨论
2.1 模型建立
采用Materials Studio 7.0软件中的Dmol3模块绘制S8、·S6·、·S4·结构,并对其结构进行基于密度泛函理论的结构优化,优化结果如图1所示。
从图1(a)可知,硫黄S8的分子结构呈皇冠对称形,S—S键的键长约为2.091 nm,键角约为108.770°,模拟得到的S8分子结构与文献值[16]一致,表明模拟体系选择合适,模拟参数设置具有可靠性。以下各模拟分子如未特别说明,均按此方法建立。
2.2 硫黄S8开环裂解路径
通过计算S8溶质硫黄分子不同位置S—S键断裂开环反应的能垒大小,确定S8开环裂解的反应路径。
首先计算S8开环断裂生成不同双组分的反应能垒,搜索能垒最低的最优反应路径,根据能量最低理论,反应能垒越低,反应性能越高,反应越容易发生,不同产物条件下的S8开环断裂反应能垒如表1所示。
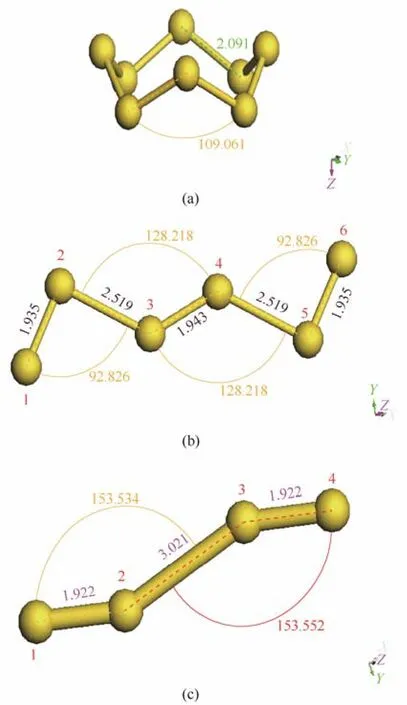
图1 S8、·S6·、·S4·分子结构Fig.1 Schematic diagram for molecular structure of S8、·S6·、·S4·

表1 溶质硫黄S8开环反应的反应能垒Table 1 Barrier energy of ring-open reaction of S8
从表1中可知,溶质硫黄S8不同开环方式所需要的能垒介于209~447 kJ·mol−1,与文献值[17]接近,证明了所设置的模拟计算参数的可靠性。断裂生成·S2·和·S6·的能垒最低,说明S8开环断裂时该反应极易发生,生成·S4·和·S4·的能垒最高,说明该反应较难发生,这是气态硫黄中·S4·含量较少的一个原因,因此在下一步继续计算断链反应时,选择S8断链易于生成的·S6·作为研究对象。
同S8一样,计算·S6·断裂生成不同双组分的反应能垒,不同产物条件下的S—S断裂反应能垒如表2所示。

表2 ·S6·自由基断键反应的反应能垒Table 2 Barrier energy of bond breaking of ·S6·
从表2中可以看出,·S6·自由基断裂生成·S2·和·S4·的能垒最低,生成·S3·和·S3·的能垒最高,说明该反应较难发生。从键长变化分析该现象[18],·S6·结构中由于受自由基的影响,图1(b)中S1—S2、S5—S6键的键长变短至1.935 nm,S2—S3、S4—S5键的键长变长至2.519 nm,键长变长,此处易于发生断裂,生成·S2·和·S4· 。因此在下一步继续计算断链反应时,选择·S6·自由基断裂链易于生成的·S4·作为研究对象。模拟计算·S4·断裂生成不同双组分的反应能垒,不同产物条件下S—S断裂反应能垒如表3所示。
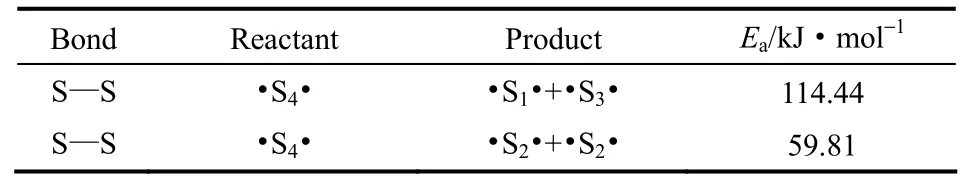
表3 ·S4·自由基断键反应的反应能垒Table 3 Barrier energy of bond breaking of ·S4·
从表3中可知,·S4·自由基断裂生成·S2·和·S2·的能垒最低,生成·S1·和·S3·的能垒最高,说明该反应较难发生。同样从键长变化理论分析该现象,·S4·结构中受自由基的影响,图1(c)中S1—S2、S3—S4键的键长变短至1.922 nm,S—S原子间作用力加强,S2—S3键的键长变长至3.021 nm,键长变长,此处易于发生断裂,生成·S2·和·S2·,使得·S4·含量非常少。
上述模拟计算过程表明1000 K以内气态硫黄分解最易生成的基本结构是·S2·,这与质谱分析的文献结果一致[19],气态硫黄S8在高温下首先分裂成·S2·和·S6·,·S6·分裂成·S4·和·S2·,·S4·继续分裂成两个·S2·,建立了反应能垒最低的硫黄裂解反应路径。

对于上述基元反应,各过渡态结构如图2所示,各过渡态的唯一虚频值见表4。
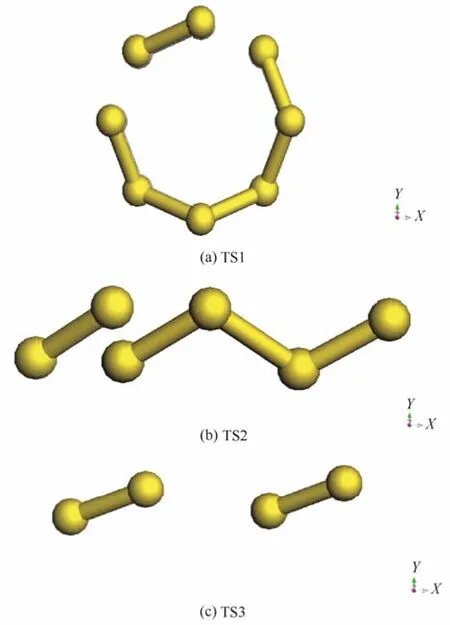
图2 各基元反应过渡态结构Fig.2 Transition state structure of each elementary reaction

表4 各过渡态虚频值Table 4 Imaginary frequency of each transition state
2.3 反应路径验证
根据化学反应过渡态理论,模拟计算各基元反应的活化能和不同温度下反应速率常数值[20],见表5。

表5 各基元反应活化能和不同温度下速率常数值Table 5 Activation energy and rate constant of each elementary reaction
建立硫黄裂解最优反应路径各基元反应的动力学方程组。
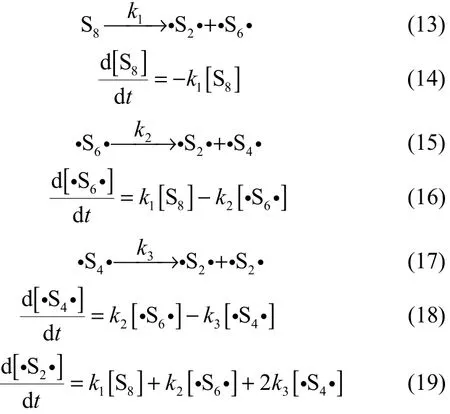
假设反应初始t=0时,设定浓度CS8=1 mol·m−3,其余组分均为0[21],反应过程设为密闭体系,固定体积,初始压力P0为101325 Pa,这与实际反应条件相一致,采用Runge-Kutta法编程计算在不同温度下各组分浓度随时间的动态变化,结果如图3所示。
分析图3可知,各组分浓度随时间变化,最后趋于稳定。在700、750、800℃温度下,·S4·的含量几乎均为0,说明·S4·极不稳定,能快速断裂成·S2·,这也从理论上验证了气态硫黄中·S4·含量极少。通过模拟计算得到的产物分布与文献实验结果[22]的对比如表6所示。

表6 组分含量模拟值与测定值比较Table 6 Comparison of simulation results and experiment results(P0=101325 Pa,constant volume)
分析表6可知,模拟值与文献测定值在3个不同温度下S8的相对误差值较大,分析原因是模拟计算时,S8自动设置为气体状态,在实验中则是硫黄在高温下首先气化,同时部分已气化硫黄处于不稳定状态,可能会重新液化,另外有部分液态硫黄发生开环聚合反应生成不溶性硫黄,因此S8含量的准确测定较为困难。在模拟计算时,自动忽略了固体硫黄液化、气化、聚合等过程,直接假设只有热裂解反应的发生,故模拟值与实验值的差别较大。其余组分的含量模拟值与实验值误差较小,体现了所建立的硫黄开环裂解反应路径的合理性,为进一步研究不溶性硫黄分子结构及聚合机理提供了参考方向。

图3 各组分浓度随温度变化模拟值(P0=101325 Pa,恒容)Fig.3 Simulation result of component concentration varied with temperature (P0=101325 Pa, constant volume)
3 结 论
(1)采用基于密度泛函理论的量子化学方法确定了在气化法制备不溶性硫黄过程中气态硫黄开环断裂反应的机理,根据能垒最小理论,模拟计算了可能的反应路径:,理论证实了气态硫黄中组分主要为·S2·、·S4·、·S6·、S8,气态硫黄中,最易生成的结构单元为·S2·。
(2)通过过渡态理论模拟计算,得到气态硫黄在开环断裂过程中各基元反应的活化能和反应速率常数,进一步通过Runge-Kutta法编程计算不同温度下(700、750、800℃)产物分布,与文献结果相符,表明了所假设的气态硫黄开环裂解反应机理的合理性,为进一步研究不溶性硫黄分子结构及聚合机理提供了参考方向。
[1] Wang Zhixia(王志霞), Chen Mingcai(陈鸣才), Liu Hongbo(刘洪波). Production technology and development of insoluble sulfur [J].Modern Chemical Industry(现代化工), 2004, 24(2): 19-22.
[2] Wang Yong(王勇), Wu Wenliang(武文良), Cao Chongyu(曹崇余), Wang Xionghui(王雄辉), Zhou Lijin(周立进). Study on the preparation and thermal stability of insoluble sulfur [J].Speciality Petrochemicals(精细石油化工), 2006, 23(2): 27-30.
[3] Ouyang Fusheng(欧阳福生), Gu Juan(顾娟), Zhang Yu(张宇), Weng Huixin(翁慧新). Study on new extractant for insoluble sulfur [J].Spciality Petrochemicals(精细石油化工), 2008, 25(3): 7-12.
[4] Jones R O, Ballone P. Density functional and Monte Carlo studies of sulfur(Ⅰ): Structure and bonding in Snrings and chains(n=2—18) [J].J. Physical Chemistry, 2003, 118(20): 9257-9265.
[5] Yuan Hongjuan(袁洪娟). Mechanism on preparation of insoluble sulfur[D]. Qingdao: China University of Petroleum, 2006.
[6] Jones R O, Ballone P. Density functional and Monte Carlo studies of sulfur(Ⅱ): Equilibrium polymerization of the liquid phase [J].J. Physical Chemistry, 2003, 119(16): 8704-8715.
[7] Du Xiaoming(杜晓明), Huang Yong(黄勇), Zhang Qian(张倩). Molecular simulation of hydrogen adsorption on NaX zeolite [J].Acta Petrolei Sinica(石油学报), 2012, 28(1): 137-140.
[8] Liu Ying(刘英), Wang Fang(王芳), Tan Tianwei(谭天伟). Applications of molecular simulation in molecular in printing technology [J].Journal of Chemical Industry and Engineering(China)(化工学报), 2006, 57(10): 2257-2262.
[9] Zhu Yu(朱宇), Lu Xiaohua(陆小华), Ding Hao(丁皓), Wang Jun(王俊), Wang Yanru(王延儒), Shi Jun(时钧). Molecular simulation in chemical engineering [J].Journal of Chemical Industry and Engineering(China) (化工学报), 2004, 55(8): 1213-1223.
[10] Hu Yao(胡瑶), Yang Xiaoning(杨晓宁). Molecular dynamics simulation of interfacial properties of gold nanoparticle in ScCO2[J].CIESC Journal(化工学报), 2011, 62(2): 295-300.
[11] Ungerer P, Nieto-Draghi C, Rousseau B, Ahunbay G, Lachet V. Molecular simulation of the thermo physical properties of fluids: from understanding toward quantitative predictions [J].Journal of Molecular Liquids, 2007, 134: 71-89.
[12] Hao Yulan(郝玉兰), Zhang Hongmei(张红梅), Zhang Hanwei(张晗伟), Li Jinlian(李金莲), Zhao Liang(赵亮). Molecular simulation research on pyrolysis mechanism of butane [J].Acta Petrolei Sinica(石油学报), 2013, 29(5): 825-829.
[13] Zhang Zhaobin(张兆斌), Li Hua(李华), Zhang Yonggang(张永刚), Xu Shixing(许士兴), Chen Shuo(陈硕), Zeng Qingquan(曾清泉). Establishment and verification of free radical model for butane steam cracking [J].Petrochemical Technology(石油化工), 2007, 36(1): 44-48.
[14] Jia Jianbo(贾建波), Zeng Fangui(曾凡贵), Li Meifen(李美芬), Xie Kechang(谢克昌). Mechanism of methane formation during toluene pyrolysis using DFT calculation [J].CIESC Journal(化工学报), 2010, 61(12): 3235-3242.
[15] Fu Xiancai(傅献彩), Shen Wenxia(沈文霞), Yao Tianyang(姚天扬). Physical Chemistry(物理化学)[M].4th ed. Beijing: Higher Education Press, 1990: 798-812.
[16] Zou Jianping(邹建平), Chen Keqian(陈克潜).Organic Sulfur Chemistry (有机硫化学) [M]. Suzhou: Suzhou University Press, 1998: 20-21.
[17] Gao Peng(高鹏). Research on production process of polymeric sulfur [D]. Shanghai: East China University of Science and Technology, 2009.
[18] Yu Ning(于宁), Long Jun(龙军), Zhou Han(周涵), Ma Aizeng(马爱增), Dai Zhenyu(代振宇), Zhao Xiaoguang(赵晓光), Zhao Yi(赵毅). Molecular simulation of dehydrogenation ofn-heptane to produce olefins [J].Acta Petrolei Sinica(石油学报), 2013, 29(2): 181-185.
[19] Cong Puzhu(从浦珠), Su Keman(苏克曼). Handbook of Analytical Chemistry(分析化学手册)[M].2nd Ed. Beijing: Chemical Industry Press, 2000: 665-667..
[20] Zhang Hongmei(张红梅), Zhang Hanwei(张晗伟), Gu Pingping(顾萍萍), Zhao Liang(赵亮). Molecular simulation research on pyrolysis mechanism of isobutane [J].CIESC Journal(化工学报), 2012, 63(10): 3138-3143.
[21] Huang Huajiang(黄华江). Practical Computer Simulation of Chemical Processes(实用化工计算机模拟)[M]. Beijing: Chemical Industry Press, 2004: 86-88.
[22] Li Zhengxi(李正西). Investigation report of insoluble sulfur [J].DDC Journal(化工开发与设计), 2000, 1: 32-42.
Ring-open reaction mechanism of sulfur S8based on density functional theory
WANG Rongjie1,2, SHEN Benxian1, MA Jian1, ZHAO Jigang1
(1State Key Laboratory of Chemical Engineering,East China University of Science and Technology,Shanghai200237,China;2Key Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan,School of Chemistry and Chemical Engineering,Shihezi University,Shihezi832003,Xinjiang,China)
Quantum chemical method based on density functional theory (DFT) was used to study the ring-open reaction mechanism of sulfur during the preparation of insoluble sulfur by gasification method. The probable reaction path was built. By the transition state theory, the activation energy and reaction rate constant were calculated for each elementary reaction. The kinetics of each elementary reaction was calculated to determine the product composition of sulfur ring-open reaction. As the result showed that the ring-open reaction of S8generated ·S2· and ·S6· preferentially, the reaction barrier was about 209.45 kJ·mol−1. The main product of S8ring-open reaction were ·S2·, ·S4· and ·S6·. The calculated values were in agreement with the experiment results based on steam density method. The result provided reference for the study of molecular structure and polymerization mechanism of insoluble sulfur.
insoluble sulfur; ring-open; reaction mechanism; density functional theory; numerical simulation
ZHAO Jigang, zjg@ecust.edu.cn
10.11949/j.issn.0438-1157.20150274
O 641
:A
:0438—1157(2015)10—3919—06
2015-03-09收到初稿,2015-04-25收到修改稿。
联系人:赵基钢。
:王荣杰(1980—),女,博士研究生。
中石化科技开发项目(LQJS1109QT0005)。
Received date: 2015-03-09.
Foundation item: supported by the Sinopec Scientific and Technological Development Projects(LQJS1109QT0005).
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
中成药(2018年9期)2018-10-09
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
橡胶工业(2015年10期)2015-08-01
橡胶工业(2015年4期)2015-07-29
