
石家庄市新生儿听力及耳聋基因联合筛查结果分析
2015-02-08 07:25封纪珍李素芳李天洁肖会者梅志勤
河北医科大学学报 2015年11期
封纪珍,李素芳,李天洁,肖会者,梅志勤
(1.河北省石家庄市妇幼保健院优生遗传科,河北 石家庄 050051;2.河北省石家庄市妇幼保健院功能科,河北 石家庄 050081;3.河北省石家庄市第五医院检验科,河北 石家庄 050021)
·论 著·
石家庄市新生儿听力及耳聋基因联合筛查结果分析
封纪珍1,李素芳2*,李天洁1,肖会者1,梅志勤3
(1.河北省石家庄市妇幼保健院优生遗传科,河北 石家庄 050051;2.河北省石家庄市妇幼保健院功能科,河北 石家庄 050081;3.河北省石家庄市第五医院检验科,河北 石家庄 050021)
目的通过分析石家庄市新生儿听力和耳聋基因联合筛查数据,探讨本区域新生儿听力筛查与耳聋基因筛查之间的关系,及早发现耳聋并对高危个体进行生活和用药指导。方法新生儿出生后3d采用耳声发射技术和自动听性脑干诱发电位两步筛查法进行听力筛查,同时采足跟血3滴应用荧光定量PCR法对遗传性耳聋常见3个基因6个位点GJB2(235delC、299-300delAT)、SLC26A4(IVS7-2A>G、2168A>G)和mtDNA12SrRNA(1555A>G、1494C>T)进行检测。听力筛查初筛未通过者需在42d进行复筛,3个月后对听力复筛未通过者及耳聋基因携带者召回进行常规听力检测。结果10 948例新生儿中9 563例通过听力初筛,初筛通过率87.35%(9 563/10 948)。1 385例未通过听力初筛的新生儿中,于42d复筛287例,复筛率20.72%(287/1 385);246例新生儿通过复筛,复筛通过率85.71%(246/287);41例未通过复筛,除去未来复筛的1 098例新生儿,最终听力筛查检出率0.42%(41/9 850)。10 948例新生儿中共检出438例携带有耳聋基因突变,耳聋基因突变携带率4.00%(438/10 948)。41例未通过听力复筛的新生儿中7例携带耳聋基因突变,携带率为17.07%(7/41);除去未来复筛的1 098例新生儿,9 809例新生儿通过了听力筛查,其中耳聋基因携带399例,携带率4.07%(399/9 809)。将耳聋基因携带率在听力筛查通过组与未通过组间比较,差异有统计学意义(P<0.05)。除去未来复筛的1 098例新生儿,9 850例新生儿中共有406例耳聋基因携带者,人群携带率4.12%(406/9 850),将耳聋基因携带率在听力筛查通过组与随机人群组间比较,差异无统计学意义(P>0.05)。最终未通过听力复筛且携带耳聋基因突变的7例新生儿中有3例235delC杂合突变,2例IVS7-2A>G杂合突变,1例IVS7-2A>G纯合突变和1例IVS7-2A>G/2168A>G复合杂合突变的患儿已确诊听力损失。结论未通过听力筛查的新生儿耳聋基因携带率显著高于通过听力筛查的新生儿;通过听力筛查的新生儿与随机人群的耳聋基因携带率差异无统计学意义。听力及耳聋基因联合筛查可以提前发现耳聋,使耳聋患儿早诊断早治疗;对高危个体进行生活和用药指导,可避免或减少耳聋的发生;还可为当地政府和医疗部门大力宣传听力及基因联合筛查的重要意义提供依据。
听力检查;婴儿,新生;基因检测doi:10.3969/j.issn.1007-3205.2015.11.009
随着新生儿听力筛查工作的开展,医务工作者们发现一些耳聋最初都是可以通过听力筛查诊断出来的[1],2007年中国人民解放军总医院聋病分子诊断中心在国际上率先提出了在新生儿听力筛查中融入聋病易感基因筛查实施联合筛查的新理念与模式[2]。本研究回顾性分析在石家庄市妇幼保健院出生并进行听力及耳聋基因联合筛查的10 948例新生儿检测结果,探讨联合筛查的重要意义,旨在对异常情况早发现、早干预,并对患儿及家属进行生活指导,以减少听障患儿的出现。
1 资料与方法
1.1 研究对象 选择2014年1月—2015年8月在石家庄市妇幼保健院出生进行听力及耳聋基因联合筛查的新生儿10 948例,男性5 713例,女性5 235例。
1.2 方法 新生儿出生后3d应用丹麦MedsenAccuscreen筛查仪进行耳声发射技术和自动听性脑干诱发电位听力筛查,仪器显示结果为“pass”或“refer”pass说明新生儿耳蜗、听神经传导通路、脑干的功能状态正常,初筛未通过者需在42d进行复筛。耳聋基因筛查于新生儿出生后3d采足跟血3滴滴到专用滤纸片上自然晾干,每个血斑直径不<8mm,应用济南英盛耳聋基因试剂盒在ABI7300实时荧光定量PCR仪上对遗传性耳聋常见3个基因6个位点GJB2(235delC、299-300delAT)、SLC26A4(IVS7-2A>G、2168A>G)和mtDNA12SrRNA(1555A>G、1494C>T)进行检测,杂合或纯合突变携带者样本均行Sanger测序得以验证。3个月后对听力复筛未通过者和耳聋基因携带者召回进行如下相应听力诊断:①声阻抗测试,应用GSItympernometry中耳分析仪进行声导抗测试;②电生理检查,应用丹麦MedsenICSCHART-EP听觉诱发电位仪进行包括短声脑干诱发电位、短纯音诱发电位和多频稳态诱发电位在内的电生理检查;③影像学检查,颞骨薄层CT扫描检查内耳、耳蜗是否畸形,MRI检查听神经发育情况;④主观听力检测,应用GSI-61双通道听力计,0~6个月采用行为观察反应,6个月~2岁采用视觉强化反应。
1.3 统计学方法 应用Excel表进行携带率的统计,计数资料比较采用χ2检验。P<0.05为差异有统计学意义。
2 结 果
2.1 新生儿听力及耳聋基因联合筛查结果 10 948例新生儿中9 563例通过听力初筛,初筛通过率87.35%(9 563/10 948);1 385例未通过听力初筛的新生儿中,于42d复筛287例,复筛率20.72%(287/1 385);246例新生儿通过复筛,复筛通过率85.71%(246/287);41例未通过复筛,除去未来复筛的1 098例新生儿,最终听力筛查检出率0.42%(42/9 850)。10 948例新生儿中共检出438例携带有耳聋基因突变,耳聋基因突变携带率4.00%(438/10 948)。41例未通过听力复筛的新生儿中7例携带耳聋基因突变,携带率为17.07%(7/41);除去未来复筛的1 098例新生儿,9 809例新生儿通过了听力筛查,其中耳聋基因携带399例,携带率4.07%(399/9 809)。将耳聋基因携带率在听力筛查通过组与未通过组间比较差异有统计学意义(P<0.05)。除去未来复筛的1 098例新生儿,9 850例新生儿中共有406例耳聋基因携带者,人群携带率4.12%(406/9 850)。最终未通过听力复筛且携带耳聋基因突变的7例新生儿中有3例235delC杂合突变,2例IVS7-2A>G杂合突变,1例IVS7-2A>G纯合突变和1例IVS7-2A>G/2168A>G复合杂合突变的患儿已确诊听力损失。见表1,2。

表1 听力筛查通过组与未通过组间耳聋基因携带情况比较
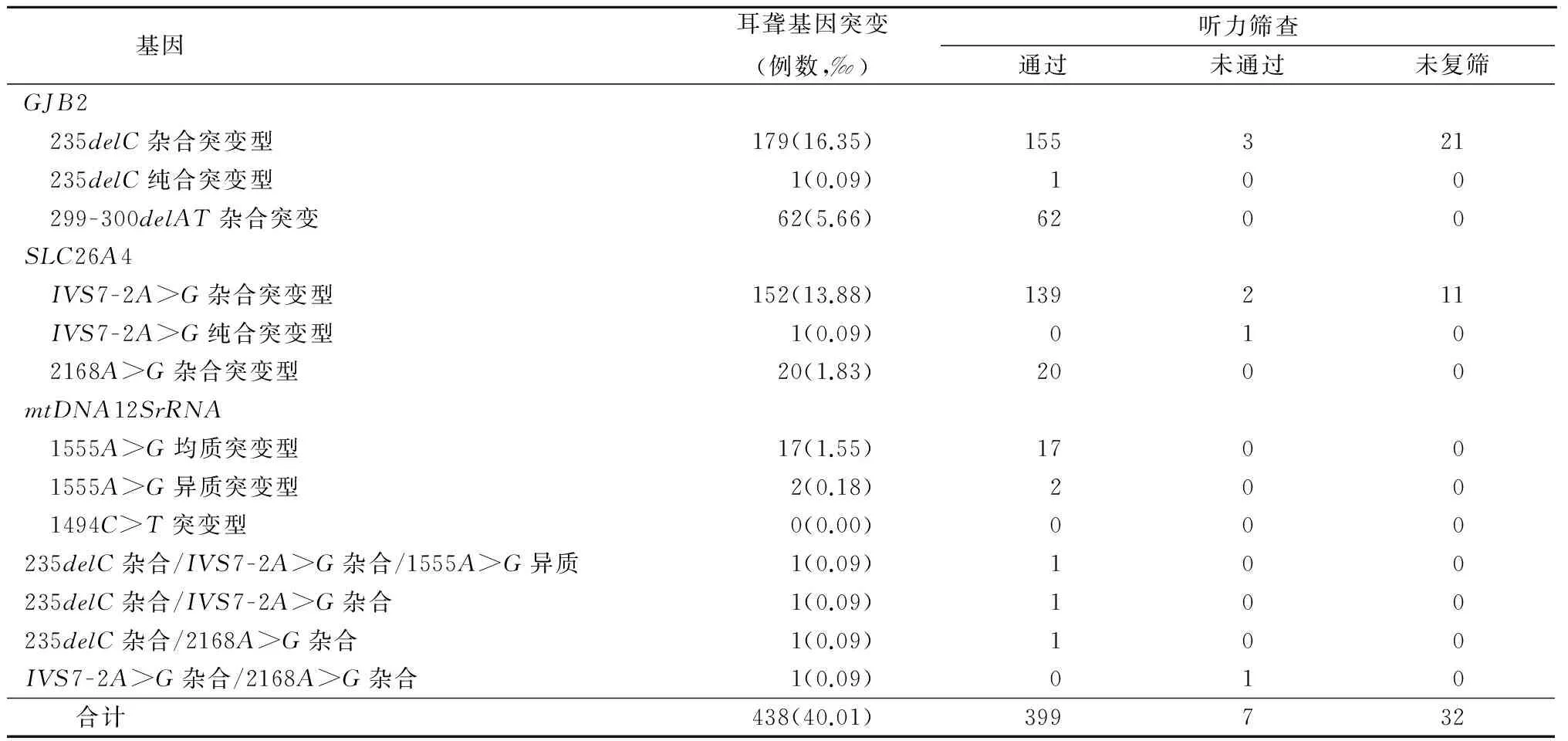
表2 10 948新生儿听力筛查和耳聋基因突变情况
2.2 特殊耳聋基因突变携带者的听力筛查及随访结果 本研究发现235delC纯合突变、235delC/IVS7-2A>G/1555A>G多杂合突变、235delC/IVS7-2A>G和235delC/2168A>G双杂合突变携带者各1例且均通过了听力筛查;1例IVS7-2A>G纯合突变携带者和1例IVS7-2A>G/2168A>G双杂合突变携带者未通过听力筛查,最终确诊该IVS7-2A>G纯合突变及IVS7-2A>G/2168A>G双杂合突变携带者为大前庭导水管扩张综合征伴耳蜗发育不全。见表3。
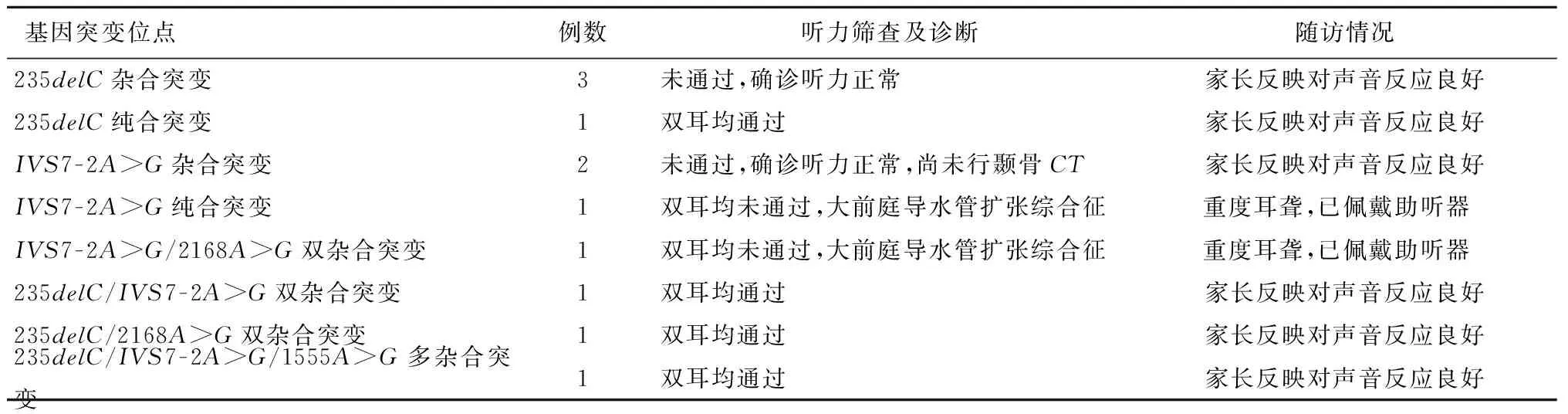
表3 特殊耳聋基因突变携带者的听力筛查及随访结果
3 讨 论
3.1 新生儿听力及耳聋基因联合筛查现状 本院2014年1月—2015年8月共出生18 450例新生儿,其中17 990例进行了听力筛查,筛查率达97.51%(17 990/18 450);10 948例新生儿同时进行了耳聋基因筛查,筛查率59.34%(10 948/18 450)。10 948例新生儿中9 563例通过听力初筛,初筛通过率87.35%;1 385例未通过听力初筛的新生儿中,于42d复筛287例,复筛率20.72%;其中246例新生儿通过复筛,复筛通过率85.71%;41例未通过复筛,最终听力筛查检出率0.42%;截至新生儿出生3月龄时仍有1 098例未来复筛,缺失率较高,相应结果存在一定偏差。由上可知,多数父母自觉患儿对声音反应良好常不予复诊,新生儿听力及耳聋基因联合筛查尚未被当地居民广泛接受。未通过联合筛查或未通过听力筛查但常见耳聋基因筛查通过者需进行进一步的听力学诊断和基因诊断,从而可有效降低耳聋发病率并避免因聋致哑的发生。本次筛查情况可作为当地相关部门制定政策及开展宣传工作提供依据。
3.2 新生儿听力及耳聋基因联合筛查结果讨论 本研究结果显示,最终41例新生儿未通过听力筛查,单纯听力筛查检出率仅有0.42%(41/9 850),耳聋基因人群携带率为4.00%(438/10 948)。表明联合筛查可以更早地发现先天性耳聋患儿或迟发型听力损失高危儿,耳聋基因筛查是听力筛查的有效补充[3]。本研究中未通过听力筛查组的耳聋基因突变携带率为17.07%(7/41),通过听力筛查组的耳聋基因突变携带率为4.07%(399/9 809),将耳聋基因携带率在两组间比较差异有统计学意义。说明未通过听力筛查的新生儿尤其要作为耳聋基因筛查的重点对象,从遗传学角度进一步明确病因,如果明确了基因诊断则可有针对性地进行干预和语言训练。如对于携带SLC26A4基因突变的高危儿,可根据其前庭导水管扩张情况[4]对其进行生活指导,避免头部受到外力撞击和使颅压增高的动作等诱发因素,极大程度降低迟发型耳聋的发病率;对于携带线粒体mtDNA12SrRNA基因突变的高危儿可进行生活和用药指导,避免使用氨基糖苷类药物,避免药物性耳聋的发生,提高康复率。
3.3 耳聋基因筛查结果讨论 本研究结果显示3个基因在石家庄市新生儿人群中的携带率由高到低依次为GJB2(携带率2.21%)、SLC26A4(携带率1.58%)和mtDNA12SrRNA(携带率0.17%);GJB2基因235delC(携带率1.64%)和SLC26A4基因IVS7-2A>G(携带率1.39%)为石家庄市新生儿热点突变位点。据报道,先天性耳聋在新生儿期的发病率为1‰~1.86‰[5],其中60%以上是由遗传因素导致的[6],遗传性耳聋患者中约有50%是由GJB2突变引起的[7],是先天性耳聋常见的致病基因。SLC26A4是仅次于GJB2基因突变而引起感音神经性耳聋的遗传学病因[8],该基因的不同突变均与前庭导水管扩张有直接关系[9]。有报道称,GJB2基因和SLC26A4基因突变患者在3.5岁前进行耳蜗移植会取得良好的效果[10]。因此,耳聋常见突变基因位点可作为门诊听障患者进行基因诊断及耳蜗移植适应证的重点检测项目。
3.4 特殊病例听力学诊断结果讨论 本院对7例携带耳聋基因突变且未通过听力筛查的患儿均进行了3月龄听力诊断,其中3例235delC杂合突变携带者和2例IVS7-2A>G杂合突变携带者的声导抗及电生理检查均正常,家长均反映新生儿对声音反应良好,但不排除同一基因上存在其他突变位点形成复合杂合导致迟发性耳聋的可能。目前每隔6个月对该5例新生儿回访1次,密切关注其听力变化情况。2例IVS7-2A>G杂合突变携带者的家长因新生儿年龄小未行颞骨CT检查,尚不能判断其与大前庭导水管综合征的关系;1例IVS7-2A>G纯合突变和1例IVS7-2A>G/2168A>G双杂合突变新生儿行颞骨CT检查后均发现双侧大前庭导水管扩张,耳蜗发育不全,诊断重度耳聋,目前已佩戴助听器。这也成为了位于等位基因上两不同位点的复合杂合突变会有叠加效应,从而产生纯合突变的效应的有力证据[11]。另外,本研究还发现1例235delC纯合突变新生儿、1例235delC/IVS7-2A>G/1555A>G多杂合突变、1例235delC杂合/IVS7-2A>G杂合和1例235delC杂合/2168A>G杂合新生儿均通过了听力筛查,并且随访时家长反映其对声音反应良好。因曾有GJB2在引起听力损失前会有“功能窗口期”的报道[12],对此家长也应引起重视,定期检测听力以防止迟发型耳聋的出现。虽有文献报道位于不同基因上的多位点杂合携带者患听力障碍的几率较低[13],但仍要密切关注其听力变化情况,尤其携带235delC杂合突变/IVS7-2A>G杂合突变/1555A>G异质突变的新生儿也应终身禁用氨基糖苷类药物,以避免药物性耳聋的发生或尽早发现迟发型耳聋。
综上所述,本研究总结分析了石家庄市新生儿听力及耳聋基因联合筛查的现状、结果和随访情况,阐明了联合筛查的重要意义。随着医学技术的发展,在新生儿听力和耳聋基因联合筛查的基础上应将耳聋基因检测推向婚检和孕产前检查等更广泛的领域,使其发挥更大的作用,从而有效地降低耳聋的出生率。
[1] 韩德民.新生儿听力及耳聋基因联合筛查[J].中国医学文摘——耳鼻咽喉科学,2012,27(6):290-292.
[2] 王秋菊,孙喜斌,黄丽辉.新生儿听力及基因联合筛查330问[J].北京:人民军医出版社,2012:2.
[3] 李琦,宋建敏,刘亚青,等.新生儿听力筛查未通过者的基因诊断[J].中华耳科学杂志,2014,12(1):37-40.
[4] 朱庆文,臧雯,袁永一,等.内耳畸形相关SLC26A4基因的研究[J].临床耳鼻咽喉头颈外科杂志,2012,26(1):22-26.
[5] 李倩,王秋菊.新生儿聋病易感基因筛查的研究进展[J].听力学及言语疾病杂志,2015,23(1):91-96.
[6] 朱晓燕,魏钦俊,鲁雅洁,等.耳聋分子病因学检测与临床应用研究[J].南京医科大学学报:自然科学版,2013,33(2):186-190.
[7] 王辉兵,于飞,戴朴,等.GJB2结构功能及致病机制研究[J].中华耳科学杂志,2013,11(1):131-137.
[8] 胡华梅,胡华,董艳玲,等.新生儿中常见的9个耳聋基因突变位点筛查分析[J].第三军医大学学报,2012,34(2):96-98.
[9] 要跟东,李守霞,张小芳,等.邯郸市特教学校耳聋患者SLC26A4基因突变分析[J].医学综述,2013,19(9):1669-1671.
[10]WuCM,KoHC,TsouYT,etal.Long-TermCochlearImplantOutcomesinChildrenwithGJB2andSLC26A4Mutations[J].PLoSOne,2015,10(9):1-13.
[11] 张华,陈俞,赵娟,等.2个耳聋家系中常见耳聋基因的基因型与表型相关性分析[J].临床耳鼻咽喉头颈外科杂志,2012,26(1):5-7.
[12] 崔庆佳,王国建,张媛,等.GJB2、SLC26A4基因相关耳聋儿童的听力损失特点分析[J].听力学及言语疾病杂志,2014,22(2):120-123.
[13] 周怡,刘海红,郝津生,等.15343例新生儿耳聋基因普遍筛查结果分析[J].中国听力语言康复科学杂志,2014,12(2):109-112.
(本文编辑:刘斯静)
近来发现网上出现冒充本刊名义的“河北医科大学学报官网(附设投稿邮箱:hbykdxxbbjb@163.com;并骗取作者将审稿费100元汇入个人帐号)”假网站,致使许多作者上当受骗,本刊强烈谴责这种不道德的行为,并保留追究其法律责任的权利。目前本刊惟一指定投稿邮箱为hbydxb@sina.com。
特此声明。
《河北医科大学学报》编辑部
Synchronous detection on screening of newborn hearing and deafness gene from Shijiazhuang
FENGJi-zhen1,LISu-fang2*,LITian-jie1,XIAOHui-zhe1,MEIZhi-qin3
(1.Department of Eugenics and Genetics,Shijiazhuang Maternal and Child Health Hospital,Shijiazhuang 050081,China;2.Department of Function,Shijiazhuang Maternal and Child Health Hospital,Shijiazhuang 050081,China;3.Clinical Laboratory,the Fifth Hospital of Shijiazhuang,Shijiazhuang 050021,China)
ObjectiveToexploretherelationshipbetweenhearinganddeafnessgenebyanalyzingthecombinationscreeningdatafromnewbornsinShijiazhuang.Toprovidelifeandmedicationguidanceforthehighriskindividualsanddetectdeafnessearly.MethodsOtoacousticemissiontechnology(OAE)andautoauditorybrainstemresponsewereusedininfantsandbloodsampleswerecollectedfromtheheelthreedaysaftertheirbirth.Sixmutationsfromthreegenes(GJB2(235delC,299-300delAT),SLC26A4(IVS7-2A>G,2168A>G)andmtDNA12SrRNA(1555A>G,1494C>T)weredetectedbyfluorescentquantitativePCR.Newbornswhofailedthefirsthearingscreeningshouldacceptitagain42dayslater.Diagnosticaudiologywasperformedonnewbornswhofailedhearingscreeningtwiceorcarrieddeafnessgenemutationsthreemonthslater.Results9 563of10 948casespassedthefirsthearingscreeningandthepassingratewas87.35%(9 563/10 948).287of1 385caseswhodidnotpassthefirsthearingscreeningcamebackforthesecondscreening42dayslaterandtherescreeningratewas20.72%(287/1 385).246newbornspassedthesecondscreeningandthepassingratewas85.71%(246/287).41casesfailedthesecondscreeningandthedetectionrateofhearingscreeningwas0.42%(41/9 850).438casescarriedmutationsamong10 948newbornsandthecarrierratewas4.00%(438/10 948).7casesof41newbornswhofailedthehearingscreeningfinallycarriedmutationswithacarrierrateof17.07%(7/41).In9 809newbornspassedthehearingscreening,399carriedmutationsandthecarrierratewas4.07%(399/9 809).Therewassignificantlystatisticdifferenceincarrierratebetweennewbornswhopassedanddidnotpassthehearingscreening(P<0.05).Therewere406mutationscarrierswithacarrierrateof4.12%(406/9 850)in9 850newbornsexceptthose1 098withoutrecallingresults.Therewasnostatisticaldifferencesbetweennewbornspassedthehearingscreeningandrandomsubjects(P>0.05).Therewere3casescarried235delCheterozygousmutationsand2IVS7-2A>GheterozygousmutationsandaIVS7-2A>GhomozygousmutationandaIVS7-2A>G/2168A>Gdoubleheterozygousmutationof7caseswhofailedthehearingscreeningfinally.ThetwocaseswhocarriedIVS7-2A>GhomozygousandIVS7-2A>G/2168A>Gdoubleheterozygousmutationsfinallydiagnosedtohavehearingloss.ConclusionTherewassignificantdifferenceonthecarrierrateofmutationsbetweennewbornswhopassedandwhodidnotpassthehearingscreening.Nostatisticaldifferencewasfoundbetweennewbornswhopassedthehearingscreeningandrandomsubjects.Thesynchronousdetectiononscreeningofnewbornhearinganddeafnessgenecoulddetecthearingdisordersearly.Wecandiagnoseandintervenehearingdefectsearlierforthosehearing-impairedchildrenandprovidelifeandmedicineguidancetohigh-riskindividuals.Thus,theincidenceofdeafnesswasreducedeffectively.Thestudyalsoprovidesabasisforthelocalgovernmentsandhealthdepartmentstoappealthesignificanceofcombinationscreeninginhearinganddeafnessgene.
hearingtests;infant,newborn;genetictesting
声 明
2015-09-24;
2015-10-16
封纪珍(1969-),女,河北正定人,河北省石家庄市妇幼保健院主任医师,从事新生儿疾病筛查、产前筛查、耳聋基因及遗传代谢病的筛查和遗传咨询研究。
R
A
1007-3205(2015)11-1271-05
*通讯作者
猜你喜欢
现代临床医学(2022年4期)2022-09-29
中老年保健(2021年12期)2021-08-24
昆明医科大学学报(2021年5期)2021-07-22
种子(2021年3期)2021-04-12
中华养生保健(2020年3期)2020-11-16
医学与法学(2020年3期)2020-09-18
World Journal of Clinical Cases(2019年6期)2019-04-17
中国生育健康杂志(2018年6期)2018-11-13
故事作文·高年级(2017年3期)2017-04-12
外语教学理论与实践(2016年1期)2016-06-11
