
我院药物临床试验不良事件监测的方案设计质量调查
2015-01-12 02:15郭晋敏赵稳华康长清
中国医药导报 2015年5期
张 莉 郭晋敏 舒 鹤 赵稳华 康长清
济南军区总医院药剂科,山东济南250031
我院药物临床试验不良事件监测的方案设计质量调查
张 莉 郭晋敏 舒 鹤 赵稳华 康长清
济南军区总医院药剂科,山东济南250031
目的分析济南军区总医院(以下简称“我院”)药物临床试验不良事件监测的方案设计现状,推动不良事件监测方案的规范化设计。方法调查我院2007~2013年Ⅱ~Ⅲ期项目的试验方案,对照不良事件的方案设计质量评分标准,分析不良事件设计质量的年均得分及各要素的年均得分率。并统计2012~2013年项目的不良事件设计质量中各要素的得分率以及各要素得分点的缺失率。结果我院药物临床试验不良事件监测的方案设计质量得分及各要素得分率均逐年提高。2012~2013年项目方案中,仍存在不良事件监测的关键信息设计缺失,其中缺失率>50%的得分点包括:妊娠事件、非不良事件的特殊事宜说明、不良事件的监测时间段、实验检查值的临床意义判定标准、不良事件与试验药物关联性评价方法、重要不良事件及导致受试者退出不良事件的报告要求、不良事件的源文件记录要求。结论试验方案中完善而周全的不良事件监测计划的制订是项目安全性评价质量的重要保证。通过对不良事件监测计划的各步骤细节进行探讨,提出不良事件监测方案的规范化设计。
药物临床试验;不良事件;设计质量
药物安全性一直是药物研发过程乃至上市后的临床运用中人们关注的重要问题。临床试验中监督试验药物的安全性是临床试验最重要的目标之一。不良事件的监测及管理是药物安全性评价的重要组成部分,原国家食品药品监督管理局于2003年发布了《药物临床试验质量管理规范》(GCP)[1],从试验方案的设计、实施和记录等各方面对规范不良事件的监测和管理做出了明确的规定。
药物临床试验方案是指导临床试验实施的纲领性文件,根据GCP要求,试验方案中应对不良事件的监测和管理进行详细的阐述,以指导研究者和申办者进行相应的安全性评价[2]。试验方案制订的好坏是药物临床试验是否能取得成功的决定性因素。因此,为了探索不良事件管理的方案设计质量,笔者探索并建立了不良事件的方案设计的质量评价标准。对照评价标准,回顾性分析济南军区总医院(以下简称“我院”)2007~2013年药物临床试验方案中不良事件管理的设计质量,探讨设计中的不足,旨在切实提高我院临床试验中不良事件的方案设计质量。
1 对象与方法
1.1 对象
检索2007年1月~2013年12月在我院药物临床试验机构的专业科室进行的Ⅱ~Ⅲ期药物临床试验。
1.2 方法
1.2.1 资料收集方法从我院药物临床试验机构办公室检索Ⅱ~Ⅲ期药物临床试验,调查其不良事件监测的方案设计情况,调查材料主要是试验方案和研究者手册。
1.2.2 不良事件方案设计的质量评分标准根据药物临床试验质量管理规范(GCP)及文献调研[3-5],制订药物临床试验不良事件方案设计质量的评分标准,实行百分制,分为8要素:已知的不良反应事件(4分)、不良事件严重性评价(14分)、不良事件的收集(12分)、不良事件的处理及随访(15分)、不良事件的评价标准(10分)、不良事件的报告(19分)、不良事件的记录(24分)、不良事件的分析汇总(2分)。详细评分标准见表1。
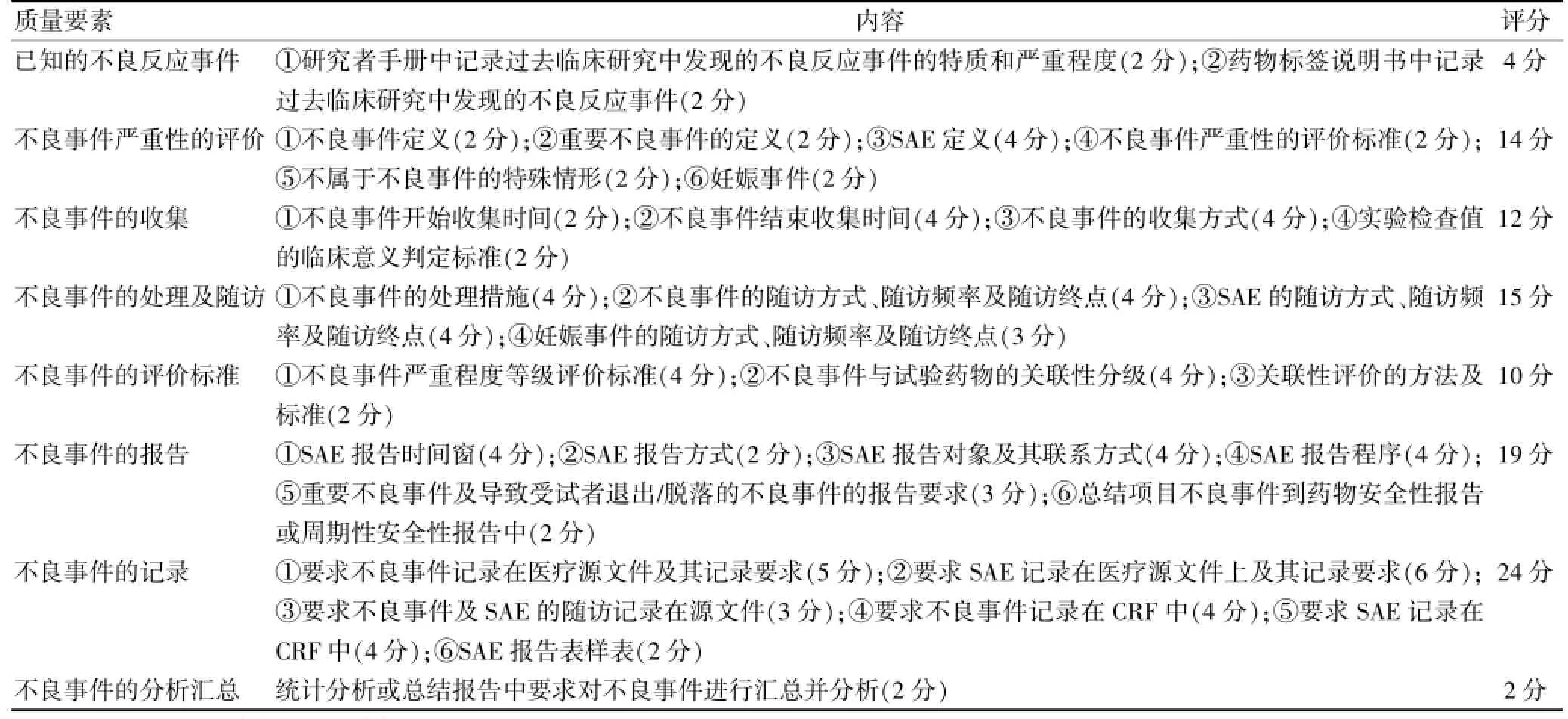
表1 不良事件监测的方案设计质量评分标准
1.2.3 观察指标①项目年均得分:对照评分标准,统计各试验项目的不良事件设计质量的得分,年均得分=年各项目的得分之和/年项目总数。②各要素的得分率和年均得分率:对照评分标准,统计试验项目中各要素的得分率(某要素的得分率=项目中某要素的得分/该要素的标准评分×100%),计算各要素的年均得分率(某要素的年均得分率=年各项目中某要素的得分率之和/年项目总数×100%)。③要素得分点的缺失率:对照评分标准,统计试验项目中各要素得分点的缺失率(要素某得分点的缺失率=缺失某得分点的项目数/项目总数×100%)。
1.2.4 指标比较①为回顾性了解我院试验项目的不良事件设计质量,统计2007~2013年各项目的不良事件设计质量的得分情况,计算项目的年均得分及各要素的年均得分率,比较不良事件设计质量的逐年变化。②为集中分析目前我院不良事件方案设计中存在的主要问题,统计近两年(即2012~2013年)项目中各要素的得分率以及各要素得分点的缺失率,以分析我院目前存在的核心问题。
2 结果
2.1 2007~2013年不良事件方案设计质量得分情况
由表2可见,我院不良事件方案设计的质量得分逐年增加。2007~2011年得分较低,位于及格线以下。从2012年开始,得分大幅度增加,至2013年已增加至80分,为2007年均得分的2倍。为进一步分析不良事件方案设计的各要点情况,对各要素的年均得分率进行统计(表3),总体来说,各要素的年均得分率亦逐年增加。截至2013年得分率不足60%的要素有1个,为不良事件的收集(54.2%)。
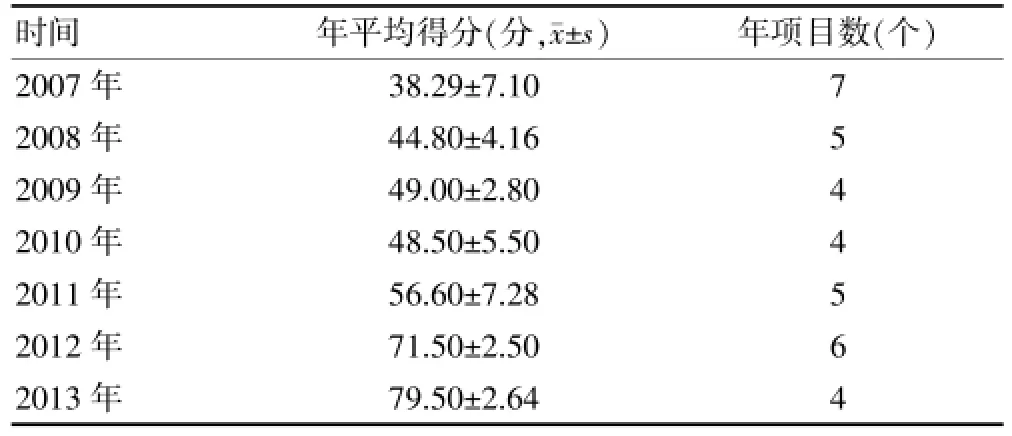
表2 不良事件方案设计年均得分
2.2 2012~2013年不良事件方案设计中各要素的质量得分情况
结合不良事件的评价标准,集中分析2012~2013年项目各要素的得分率以及各要素中质量得分点的缺失率,以评价目前我院方案设计中存在的问题。
①已知的不良事件,得分率为80%。作为试验药物的安全性信息,各项目研究者手册中均无缺失,在药物标签说明书中有40%的项目缺失该部分信息。②不良事件严重性评价,得分率为81.43%。其中,存在缺失的质量得分点为重要不良事件的定义、不良事件严重性的评价标准、非不良事件的特殊事宜说明和妊娠事件的处理要求,缺失率分别为20%、30%、30%和50%。③不良事件的收集得分率最低,为48.33%,该要素中的缺失率也最高。对不良事件的开始及结束监测时间,90%项目方案中未做明确规定。实验检查值的临床意义判定标准,即不良事件判定标准,40%项目未做明确规定。④不良事件的处理及随访,得分率为70.67%。各质量得分点(不良事件的处理措施、不良事件的随访、严重不良事件(SAE)的随访和妊娠事件的随访)均存在缺失,缺失率分别为30%、20%、20%和50%。⑤不良事件的评价标准,得分率为82%。其中,存在缺失的质量得分点为不良事件的严重程度评价标准和关联性评价方法,缺失率分别为10%和75%。⑥不良事件的报告,得分率为81.05%。各方案中均说明SAE的报告时间窗、报告方式、报告对象和报告流程,但仍有10%项目未提供报告对象的联系方式,10%项目未详细提供EDC系统的SAE报告流程。重要不良事件及导致受试者退出不良事件的报告要求和药物安全性报告/周期性安全性报告的缺失率较高,分别为70%、50%。⑦不良事件的记录,得分率为75.42%。该要素中缺失得分点集中在不良事件的源文件记录上,80%项目说明了不良事件及SAE的记录内容及要求,但70%项目对源文件记录未做明确说明。⑧不良事件的分析及汇总,得分率为100%,各项目中该要素信息完备。
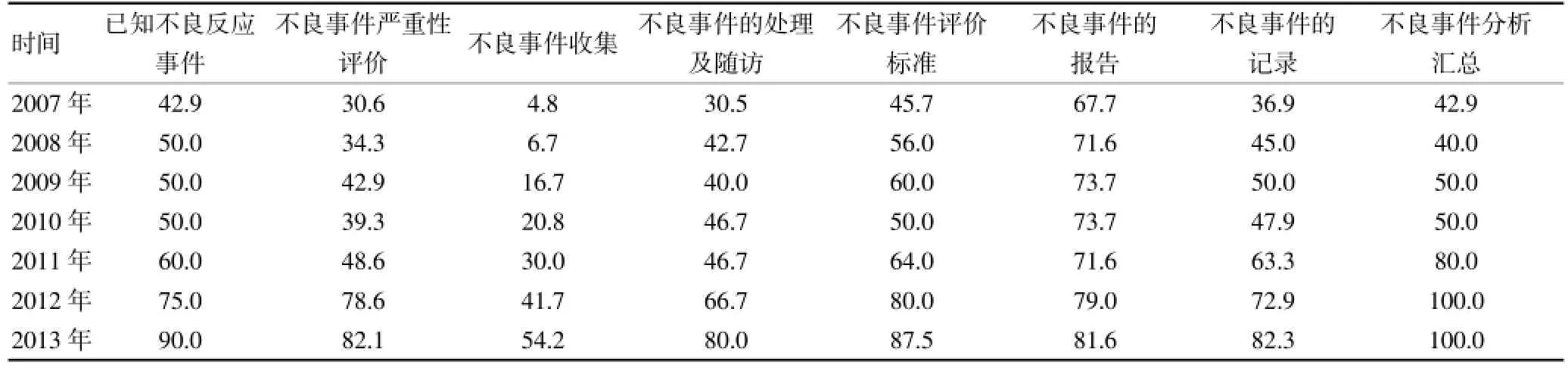
表3 不良事件方案设计各要素的年均得分率(%)
3 讨论
3.1 本次调研发现问题
本文将试验方案中不良事件的安全性监测计划分为8步:已知不良反应信息的告知、不良事件的定义告知、不良事件的收集、处理及随访、评价、报告、记录和统计分析。不良事件监测工作的每一环节均影响监测质量,申办方和研究者应充分认识到每一步骤的重要性。只有试验方案中制订完善而周全的不良事件监测计划,才能保证项目中监测工作的顺利实施及实施质量[6]。本次调研发现,近年来虽然我院项目方案中不良事件安全性监测计划的质量逐年提高,但截至2013年仍存在一些关键信息的设计缺失,而这正反映出申办方和研究者对不良事件安全性监测警戒意识较薄弱的环节。缺失信息集中体现在:方案中未对妊娠事件的处理流程、非不良事件的特殊事宜说明、不良事件的开始及结束监测时间、实验检查值的临床意义判定标准、不良事件与试验药物关联性评价方法、重要不良事件及导致受试者退出不良事件的报告要求、不良事件的源文件记录作出明确的规定(缺失率>50%)。另外,部分信息在方案中的规定不够细致及完整,如不良事件的收集方式,各项目阐述深度有差异,有的项目中为研究者提供了多样化的收集方式,以保证收集的完整性,而有的项目中只简要介绍标准化提问方式。另外,多数方案中对源文件记录内容的要求并不完整,试验项目信息和受试者鉴别信息是常见的缺失内容。
3.2 方案中不良事件监测计划的规范化设计
不良事件作为临床试验的重要安全性信息,在试验方案的安全性评价中,其监测和管理通常被单独列为一个章节,予以详尽和准确的阐述[7]。结合本次调研发现问题,通过对不良事件监测计划的各步骤细节进行探讨,为完善和规范试验方案中不良事件监测计划设计提供参考。
3.2.1 已知不良事件的告知申办者应提供该药物的临床前安全性研究资料及其他与安全性有关的资料,包括已知的不良反应,列入研究者手册或药物标签说明书中。
3.2.2 明确不良事件的定义在方案中应对涉及到的术语做出明确定义,如不良事件、重要不良事件、SAE等。对于不良事件的判定和严重性分级也应在方案中明确说明。根据不良事件的严重性,一般分为SAE、重要不良事件和一般不良事件。不同性质的不良事件,有不同的后续处理流程。方案中应尽可能给出各级不良事件的处理标准操作规程,尤其是SAE,可附录中予以说明。对不良事件的认知有时会受到研究者主观意识的干扰,因此,对不良事件作出明确的定义,对研究者更具有指导意义。对于不属于不良事件的特殊情形[8],在试验方案的安全性监督和报告部分应当予以预先定义或描述。例如,年度体检的行政性入院、在进入试验项目前就已计划或安排的手术或住院程序等“住院事件”通常不需要被当作SAE而进行监督和报告。对于妊娠是否应当被视为不良反应事件以及如何跟踪妊娠结果在试验方案中应当有明确的规定[9]。临床试验中,任何妊娠后的受试者通常应当被立即终止参加试验项目,并在获知后的第一时间里完成妊娠事件报告,上报申办者。对妊娠的后续跟踪监督视对妊娠结果的要求而定,有些到婴儿出生为止,有些需要继续跟踪婴儿的成长直至成年。
3.2.3 不良事件的收集方案中应明确规定不良事件的监测时间窗。一般对不良事件的监督从受试者签署知情同意书或第1次服药之后开始,直到他们完成或退出试验项目或最后1次服用研究药物或最后1次访视后的30 d[10]。不良事件的收集方式,应明确列在试验方案中。常见的收集方式有:建立标准化提问表、建立症状检查清单、研究者开放式提问、日志记录和同期服用药物询问。根据项目特点,尽量采用多样化收集方式,才能避免不良事件监测的遗漏。如针对长期口服试验药物的门诊受试者,可让受试者把访视期间的不良事件经历记录在受试者日记卡中,当访视时,研究者与受试者一起回顾日志信息,并当场澄清任何不良事件的疑问。同期服用药物的询问,常常也能够牵扯出新的不良事件。根据体格检查和实验室指标而收集不良事件,常常需要与试验方案规定的基线相比较[11]。因此,方案中应明确规定指标异常变动的临床意义判定标准。若判断为异常有临床意义,即为不良事件。若未进行明确规定,则需研究者根据行医经验、行业标准判断是否有临床意义。
3.2.4 不良事件的处理流程在临床试验中,一旦受试者发生不良事件,均应按照医疗常规进行积极适当的处理。方案中应根据不良事件的严重程度,给出建议的处理措施。对不良事件的处理措施一般分为:未采取措施、调整药物剂量、暂时中断试验、服用伴随药物、非药物治疗、永久停药,退出试验、住院/延长住院时间。在双盲试验中,还应明确紧急破盲的标准操作规程。研究者有责任对不良事件进行治疗或监视,跟踪直至有最终结局。试验方案中应明确不良事件及SAE的随访方式、随访频率及随访终点。需强调的是,试验结束仍未解决的SAE一般应被监督到下列情形之一出现为止:事件解决、事件稳定、事件回到基线状态、事件的继续监督变得无法完成(如与事件当事人失去联络等)。如果申办者希望执行所有这些继续监督的要求的话,就应当明确把它们列在试验方案安全性监督部分中。
3.2.5 不良事件的评价方案中应明确不良事件严重程度的等级评价标准。严重程度常分为轻度、中度和重度。亦可根据美国国家癌症研究所(NCI)《常见急性及亚急性毒性分级标准》分为4度,在NCI毒性标准未列出的不良事件,可另附等级判断标准。不良事件与试验药物的因果关系是另一个需要评价的方面,应当对试验方案予以定义,包括关联性评价的分级标准(如5级标准或7级标准)以及评价的方法(如时间挂钩法、滞后期效应法、排除法等)。不良事件与药物的因果关系常常难以判断,特别是当不良事件不符合已知的药物安全性信息时,评价标准及评价方法的确定,可使研究者有据可依,更利于评价。在临床试验中,药物不良反应的安全性问题需要引起重视和报告,因此,如何判断不良事件是否属于药物不良反应,在试验方案中应明确。在临床试验阶段,所有与试验药物及其任何剂量有关的有害和不期望的反应都被视为药物不良反应。它与研究药物有可能存在一定的内在关系,即药物与不良反应事件的直接关系不能被排除。已知的不良反应在研究者手册中已经进行说明,在试验中尤其应重视未知不良反应事件/非预期药物不良反应。申办方对药物不良反应的处理及报告要求在试验方案也应明确列出。
3.2.6 不良事件的报告流程根据GCP规定,若发生SAE,要求研究者在获知24 h内向药品监督管理部门、卫生行政部门、申办者和伦理委员会报告。对于多中心大规模临床试验的SAE,一般还需要申办方立即向参加试验的所有中心通报,以使其他研究者提高警惕。因此,在试验方案中应明确规定SAE的报告时限、报告流程、报告对象以及提供相应的联系方式[12]。此环节中需强调的是申办方若使用临床电子数据采集系统(EDC),应明确SAE报告流程。在EDC系统,研究者需要以SAE报告封面页的形式在获知SAE后的24 h内通告申办方有关SAE的发生。若EDC系统具有完全的SAE报告功能的话,申办方可在接到SAE信息通告后直接将SAE报告或表格,从EDC系统中提取出来,直接提交相应的SAE报告。如果EDC系统不具有SAE报告功能的话,研究者仍需要完成书面SAE报告,并打印和附上相关SAE表格作为支持信息,一并上报。对于重要不良事件以及导致受试者退出的不良事件,因其临床重要性,应定期向申办方报告。若大规模临床试验且持续时间较长,普通不良事件要求每季度向申办方汇报一次。诸如此类对上报的时间及频率有要求的,应在试验方案中明确规定。
3.2.7 不良事件的记录受试者在试验过程中发生的所有不良事件,均应详细记录在源文件中并转抄至病例报告表中。源文件的记录是试验实施中常见的薄弱环节[13]。明确而全面的记录要求能在一定程度上促进研究者清晰地完成不良事件的描述。首先,方案中明确源文件及其记录内容,包括试验项目信息(试验药物信息、试验方案、研究机构编号)、受试者鉴别信息(受试者随机号、姓名缩写、性别、与不良反应事件有关的病史、同期服用药物情况)、事件诊断、事件描述、严重程度、关联性、处理措施、事件结果。另外,SAE还要求记录事件上报情况(上报时间、方式及对象)。其次,指出记录注意事项,如记录不良事件到源文件中时应尽可能采用受试者的原叙述(原话)。在输入病例报告表时由研究者根据医学判断转换成相应的医学名词或术语以及相关的事件归类。
3.2.8 不良事件的统计分析在试验方案的统计分析部分,应明确指出对不良事件的统计分析策略,以评价试验药物或研究产品的总体安全性。
4 结语
安全性评价作为临床试验中与疗效评价同样重要的一个方面,应引起足够的重视。随着我国药物临床试验与国际接轨,有必要提高我国安全性评价的规范化水平。首先在临床试验准备阶段应当制订良好的项目安全性监测计划[14]。只有通过试验方案制订详细而周全的不良事件监测和管理的安全性评价计划,才能指导研究者和申办者尽可能全面地对不良反应事件的面貌予以了解,以便给患者提供足够的药物安全信息或警讯,并做好管理和预防药物风险使用的工作[15]。
[1]国家食品药品监督管理局.药物临床试验质量管理规范[S]. 2003-09-01.
[2]刘川.临床试验方法学[M].北京:化学工业出版社,2011:257-279.
[3]Prady SL,Richmond SJ,Morton VM,et al.A systematic evaluation of the impact of stricta and consort recommendations on quality of reporting for acupuncture trials[J]. PLoS ONE,2008,3(2):e1577.
[4]邢建民,李迅,刘建平.药物临床试验安全性报告的规范[J].中国药物警戒,2010,7(4):202-205.
[5]李少丽,颜敏,吴晔,等.药品临床安全性评价与药品临床试验管理规范的相关要求[J].药物流行病学杂志,2003,12(1):1-6.
[6]唐雪春,宋苹,张勋.药物临床试验机构对临床试验中不良事件的监控[J].中国新药与临床杂志,2006,25(3):228-231.
[7]任明,商洪才,张伯礼,等.临床试验中不良事件的管理[J].中国临床药理学杂志,2008,25(5):452-457.
[8]范大超.临床试验安全性通报的常见问题[J].中国处方药,2010,6(99):70-71.
[9]范大超.新药临床试验安全性通报及监测[J].中国处方药,2010,6(98):70-71.
[10]胡晓民,吴磊.临床试验中不良事件的监测和管理体会[J].广州医药,2005,36(5):66-67.
[11]袁延楠,姚晨,阎小妍,等.临床试验中实验室检测数据的可视化评价[J].中国新药杂志,2012,21(4):402-430.
[12]汪秀琴,熊宁宁.临床研究不良事件的伦理审查[J].中国新药杂志,2010,19(15):1299-1301.
[13]沈玉红,张正付,李正奇.我国药物临床试验实施问题及对策[J].实用药物与临床,2013,16(2):173-176.
[14]陆芳,高蕊,唐旭东,等.如何制定临床试验的数据和安全监察计划[J].中国临床药理学与治疗学,2010,15(5):584-587.
[15]顾俊,李娜,张志勇.浅议药物临床试验机构的风险管理[J].中国新药与临床杂志,2011,30(10):797-800.
Investigation on project design quality of adverse events in clinical drug trial protocol of our hospital
ZHANG LiGUO JinminSHU HeZHAO WenhuaKANG Changqing
Department of Pharmacy,Ji'nan Military General Hospital,Shandong Province,Ji'nan250031,China
ObjectiveTo analyze design quality of adverse events monitoring in clinical drug trial of Ji'nan Military General Hospital(“our hospital”for short)and to promote normalization design of adverse events protocol.MethodsTheⅡ-Ⅲperiod clinical drug trial protocol from 2007 to 2013 in our hospital was investigated,in comparison with score criterions of design quality for adverse events,the average scores per year and the score rate of items were analyzed.In addition,the score rate of items and the missing rate of each score point from 2012 to 2013 clinical drug trial protocol were also analyzed.ResultsThe design quality score of adverse events in clinical drug trial protocol of our hospital,as well as the score rate of items,increased year by year.However,there were still some defects in 2012-2013 clinical drug trial protocol.The missing score point of design items,with the missing rate more than 50%,were shown as follows:pregnancy events,exclusions of adverse events,monitoring time of adverse events,criteria for clinical significance of experimental test value,the methods of association evaluation between adverse events and trail drugs, reporting requirements of significant adverse events and adverse events which caused the withdraw of subjects,the source file record requirements.ConclusionEstablishing of a complete and comprehensive monitoring plan of adverse events in protocol is an important guarantee for assessment guality of project safety.By the discussion in details to each step of adverse events monitoring plan,normalization design of adverse events monitoring protocol is proposed.
Clinical drug trial;Adverse events;Design quality
R95
A
1673-7210(2015)02(b)-0156-05
2014-11-02本文编辑:张瑜杰)
张莉,女,硕士研究生,副主任药师;研究方向:药物临床试验、临床药学、药事管理。
舒鹤,博士研究生,副主任药师;研究方向:临床药学、药物临床试验。
猜你喜欢
汽车工程师(2021年12期)2022-01-18
北京航空航天大学学报(2021年9期)2021-11-02
基层中医药(2020年5期)2020-09-11
初中生世界·九年级(2017年8期)2017-09-06
信息安全与通信保密(2016年3期)2016-08-23
电子设计工程(2015年12期)2015-02-27
中国合理用药探索(2012年2期)2012-03-20
中国合理用药探索(2011年9期)2011-03-20
中国合理用药探索(2011年7期)2011-03-20
