
Myocardin in biology and disease
2015-01-10 06:16:24JosephMiano
Joseph M.Miano
Aab Cardiovascular Research Institute,University of Rochester School of Medicine and Dentistry,Rochester,NY,USA.
Myocardin in biology and disease
Joseph M.Miano✉
Aab Cardiovascular Research Institute,University of Rochester School of Medicine and Dentistry,Rochester,NY,USA.
Myocardin(MYOCD)is a potenttranscriptional coactivator that functions primarily in cardiac muscle and smooth muscle through directcontacts with serum response factor(SRF)over cis elements known as CArG boxes found near a number of genes encoding for contractile,ion channel,cytoskeletal,and calcium handling proteins. Since its discovery more than 10 years ago,new insights have been obtained regarding the diverse isoforms of MYOCD expressed in cells as wellas the regulation of MYOCD expression and activity through transcriptional, post-transcriptional,and post-translationalprocesses.Curiously,there are a number of functions associated with MYOCD thatappear to be independentof contractile gene expression and the CArG-SRF nucleoprotein complex. Further,perturbations in MYOCD gene expression are associated with an increasing number of diseases including heartfailure,cancer,acute vesseldisease,and diabetes.This review summarizes the various biologicaland pathologicalprocesses associated with MYOCD and offers perspectives to several challenges and future directions for further study of this formidable transcriptionalcoactivator.
Cell lineage determination and differentiation require context-dependentextracellular and intracellular signaling events that converge upon the nuclear genome to coordinate specific patterns of gene expression requisite for normal cellular homeostasis.Such programs of gene transcription require cell-restricted and more widely expressed DNA binding transcription factors and their attending co-regulators that fashion the epigenome forappropriate controlof gene expression.Skeletal muscle cells were the first cell type shown to arise through the activity of a single DNA binding transcription factor[1].This factor,named MYOD1,represented a paradigm of cell specification[2],and its discovery triggered a surge in interest to identify similarly acting transcription factors that could contribute to the identity of other distinct cell types such as neurons,cardiac muscle cells,beta cells of the pancreas,and various hematopoietic cells[3-5]. These and other seminalstudies further indicate that a cellˊs phenotype is notfixed;rather,underappropriate conditions,cells may undergo transdifferentiation from one celltype into another[6].
Unlike mostofthe~250 distinctcelltypes thatarise from discrete regions of the developing embryo,vascular smooth muscle cell(SMC)types originate from multiple points in the embryo and exhibit considerable phenotypic plasticity during development and disease[7-11].Throughout the 1990s and early 2000s, investigatorsutilized such techniquesaslow stringency degenerate oligonucleotide screening[12,13],mRNA differentialdisplay[14,15],and interaction cloning[16-18]to discovertranscription factors thatcould program SMC differentiation in a manner similar to MYOD1;however,none ofthe identified factorsexhibited a MYOD1-like function.A major breakthrough occurred in 2001 when Da-ZhiWang in Eric Olsonˊslaboratory discovered myocardin[19].Asdetailed below,myocardin(MYOCD) is both necessary and sufficient for the developmentand differentiation of most SMC,suggesting ithassome features in common with MYOD1.The purpose ofthis review is to summarize the initialfindings related to MYOCD as a powerfulco-activatorof serum response factor(SRF)-dependent gene expression in cardiac muscle and SMC and the subsequentliterature on its expression,regulation,and function in molecular and pathologicalprocesses thatextend beyond the cardiovascularsystem.Partone summarizestheinitialdiscovery,mRNA expression,and majorfunction of MYOCD asa vitalcomponentto a molecularswitch for SMC differentiation.Part two addresses the transcriptional, post-transcriptional,and post-translationalcontrolof MYOCD as wellas the growing numberof regulatory proteinsthatalteritsexpression oractivity.In partthree, the role of MYOCD in otheraspects of cellbiology is synopsized including suppression ofcellgrowth,modulation of microRNAs and ion channels,repression of skeletal muscle differentiation,and several CArG-independentfunctions.Partfourwillhighlighta number ofpathologicaldisorders in which MYOCDis suspected to play some role.Throughout each section,future directions for the study ofthis amazing co-activatorof gene expression are offered.
Discovery,expression,and initial characterization of myocardin function
The discovery of MYOCD is a lesson in applying computationalbiology to gene discovery.Exploiting the wealth of data accumulating from globalwork on transcriptomics,Wang etal.used the simple BLAST search algorithm to compare expressed sequence tags from embryonic cardiac muscle cDNA libraries with existing sequence data and found one of the 20 novel sequences to correspond to the 3ˊuntranslated region of MYOCD[19].Myocardin was so named because of its primarily cardiac mu scle-restricted pattern of expression during embryogenesis and in the adult[19]. Initialconstruction ofthe mouse Myocd cDNA revealed an open reading frame of 807 amino acids with the amino-terminal300 amino acids consisting of a basic domain,a SAP domain common to transcription factors mediating changes in genomic architecture,and a polyglutamine domain found commonly in transcription factors.Interestingly,expansion of polyglutamine domains in certain transcription factors is implicated in neurodegenerative diseases[20]though such augmentation of polyglutamine tracts in the human MYOCD gene have not been reported in neurons.The carboxyterminaldomain of MYOCD shows little homology to other proteins except for a leucine zipper motif that appears to mediate MYOCD homodimerization[21]. Nuclearlocalization ofectopically expressed MYOCD and GAL4 reporter assays supported the idea that MYOCD exhibits transcriptionalactivity[19].Deletion analysis indicated thatmostofthe transcriptionalactivity of MYOCD ismediated through the carboxy-terminal 300 amino acids.Reporter assays confirmed MYOCD to be a strong transactivatorofpromoterscontaining CArG boxes,which are found predominantly in muscle and cytoskeletal genes[22,23].Depending upon context,the degree of MYOCD transactivation was more than three orders of magnitude above baseline making MYOCD one of natureˊs titanic transcriptional co-activators. Finermapping studies showed thatthe basic and polyglutamine domain were essentialforMYOCDtranscriptional activity.Gel shift assays demonstrated MYOCD was unable to bind to a CArG box from the Tagln promoter;however,MYOCD wasshown to physically bind serum response factor(SRF),a ubiquitously expressed DNA binding transcription factor[24],and together SRFMYOCD formed a ternary complex overthe Tagln CArG box[19].Apparently,MYOCD does not directly contact DNA,though X-ray crystallography experiments of MYOCD bound to SRF over a CArG box should be done to formally prove this point.The association of MYOCD with SRF requires the basic and polyglutamine domains of MYOCD coming into contactwith the amino-terminal MADS domain of SRF[19].The results ofthis elegantseries ofexperiments proved that MYOCD,like many transcription factors,is modular with respect to its functionality;the amino-terminus serves to bind to SRF and the carboxy-terminus mediates strong transcriptionalactivation.Deletion of the carboxy-terminal domain of MYOCD resulted in a dominantnegative protein that could,if ectopically expressed in developing Xenopus embryos,inhibit endogenous cardiac muscle gene expression[19].This finding suggested that MYOCD was a criticalcofactor forthe cardiac muscle program and mightbe necessary fornormalcardiogenesis.Two othermyocardin-related factors were subsequently discovered and have been reviewed elsewhere[25-28].
Expression profile of myocardin
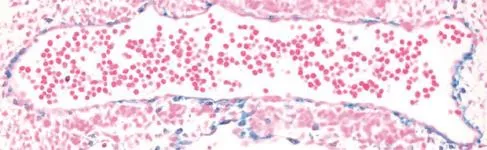
Fig.1Lineage tracing of myocardin in developing mouse aorta.Beta galactosidase staining(blue SMC)ofembryonic day 10.0 mouse aorta. Result was obtained in embryos carrying the R26R reporter gene and Cre recombinase knocked into the endogenous Myocd locus.
Initial Northern blotting showed Myocd mRNA to be restricted to adulthearttissue and in situ hybridization of developing mouse embryos confirmed robust expression in the cardiac crescentas early as embryonic day 7.75[19].Levels of Myocd mRNA persistin the developing mouse heart over later stages of development and could also be detected in SMC of gutand vasculature[19,29].Myocd mRNA expression could not be detected in skeletal muscle,either in vivo or in vitro[19,30].Further,in situ hybridization failed to detect Myocd mRNA in vascular SMC prior to embryonic day 11.5 in the mouse[29].Lineage tracing data,however, suggestthat Myocd is presentin the dorsalaorta as early asembryonic day 10.0 ofdevelopment[31](Fig.1).This suggests that Myocd mRNA may existin early vascular SMC lineages,butis below the levelofdetection using standard methods.Expression of Myocd mRNA in heart and SMC-containing tissueswasconfirmed in developing Xenopus[32]and chicken[33].RNase protection assay showed robustexpression of Myocd mRNA in adultrat aortic SMC,with levels approximating those seen in adulthearttissue[30].More extensive mRNA profiling revealed human and mouse Myocd mRNA are expressed highly in many adultSMC-containing tissues[29,34].Myocd mRNAisnotdetectable in endothelialcellsbutisinduced in a multipotent stem cell line stimulated to become SMC[34].Moreover,Myocd mRNA iselevated following stimulation with all-trans retinoic acid[35](a potentphenotypic modulator of SMC in vitro and in vivo[36]), potassium chloride[37],stretch[38],thrombin[39],NOX4[40], NRF3[41],and TGFβ1[42].As discussed below,levels of MYOCD mRNA are modulated in association with various human diseases.While many commercialantibodies to MYOCD are available,they do notappear to reliably detect endogenous MYOCD protein,perhaps due to low-levelexpression.Thus,untila more widely accepted and validated antibody to MYOCD is developed,assessing endogenous MYOCD willbe largely restricted to mRNA levels.Utilization of proximity ligation assay or knocking in an epitope tag(such as 3xFLAG)into the endogenous mouse Myocd locus could circumvent challenges presented with existing antibodies to this cofactor.
Myocardin is an essential cofactor for the SMC differentiated phenotype
SMC are notorious forexhibiting a range ofphenotypes both in vitro and in vivo[10].In general,the contractile phenotype of SMC is reduced when they are cultured orfollowing injury to the vesselwall.Thisprocess was originally described as de-differentiation[43],but is more often referred to as phenotypic modulation[44], phenotypic plasticity[45]or phenotypic switching[46]. For decades,the transcriptional processes associated with this change in SMC phenotype remained a mystery.The initialreporting of MYOCD acting with SRF to strongly induce cardiac gene expression[19]launched an effortto define the role of this factor in SMC.The first preliminary study in early 2002 indicated that MYOCD could serve a MYOD1-like role in SMC[47]. A subsequentpaperpublished in the same yearreported fournovelfindingspertaining to MYOCD and the SMC differentiation program[30].First,levelsof Myocd mRNA were shown in an RNase protection assay to be lower in immortalized or primary-derived cultures of SMC as compared to SMC within aortic tissue.This result suggested that MYOCD is partofthe biochemicaldedifferentiation program that occurs when SMC are removed from theirnative milieu and induced to proliferate in a culture dish.Second,overexpression of MYOCD induced the activity of severalSMC promoters in a luciferase assay,including the highly specific promoter, Myh11.Severalother SMC-restricted genes were later shown to exhibit MYOCD-dependentexpression and promoter activity including the potassium channel, Kcnmb1[48],Lmod1[49],and telokin[50].Third,ectopic expression of MYOCD in a cellline thatexhibits no detectable expression of Myocd mRNA resulted in the stimulation of SMC marker genes such as Acta2 and Cnn1.This MYOD1-like gain-of-function study was the firstto clearly demonstrate a role for MYOCD in directing an endogenous SMC differentiation program. Finally,overexpression of MYOCD could bluntcell growth,a key characteristic ofdifferentiated,contractile SMC in the vesselwall.Collectively,these findingsthe reduced expression of Myocd mRNAin de-differentiated SMC,MYOCD-induced activation of SMC promoters and endogenous SMC marker genes,and the attenuated growth of cells with overexpression of MYOCD-supported a new concept for MYOCDfunction,namely,itsprimacy in directing a SMCdifferentiation program[30].These findings were subsequently confirmed and extended in importantways by several independent groups[21,29,34].MYOCD also appears sufficientfor directing a functionally competent SMC phenotype based on ultrastructuraland physiological experiments(e.g.,calcium flux and slow wave contraction)[51-53].Interestingly,there is in vivo evidence to support ectopic MYOCD converting hepatocytes to a SMC-like state[54].Thus,whereas the transdifferentiation ofa celltype into anotherdistinctcelltype often requires a cocktail of transcription factors[5,55],it appears that ectopic expression of a single factor(i.e.,MYOCD)is adequate for the conversion of a non-SMCinto an SMC-like phenotype.Whether cells transduced with MYOCD first revert to a pluripotency state before differentiating into SMC is unclear.Collectively,in vitro studies have established MYOCD as the principal mediator of the normalvascular SMC contractile phenotype.
Further evidence for MYOCD functioning to direct the SMC differentiation program comes from mouse genetic studies.Wang et al.showed firstthata panknockout of Myocd resulted in mid-gestation arrest with little evidence ofaortic SMC differentiation;interestingly,however,embryonic vessels were patterned normally and the heartdid notappear to display any obvious defect,though there were signs of defects in the yolk sac vasculature[56].Subsequentstudiesindicated a strong heart phenotype in mice lacking normal MYOCD[57,58].The reasons for such disparate cardiac data are unclear,butcould relate to the strain ofmouse studied.Itisalso relevantto pointoutthatevidencesupports some SMC differentiation in vessels where MYOCD is knocked out,suggesting thatparallelpathwaysexistto directthe SMC differentiation program[59]. The weight of evidence,however,clearly supports a dominantrole for MYOCD in the establishmentand maintenance ofa SMC contractile phenotype in vivo[60].
Regulatory control of myocardin expression and activity
Several levels of regulation in MYOCD expression and activity have been studied over the last 10 years, and these can be broadly defined in terms oftranscriptional,post-transcriptional,and post-translational control processes.Transcriptional processes include promoter studies and the identification of cis-acting elements either activating orinhibiting gene transcription.Post-transcriptionalprocesses encompass splicing and stability with the latter governed by a growing number of microRNAs.Post-translational processes involve various modifications to the MYOCD protein (e.g.,phosphorylation)that modulate its expression or activity.Each of these control processes will be reviewed briefly next.
Transcriptional control of myocardinexpression
Severalin vitro studies have defined regulatory elements controlling Myocd promoter activity and,by extension,Myocd mRNA expression.The firstpositiveacting elementdescribed wasa proximalbinding site for the cardiac muscle-restricted transcription factor, NKX2-5[61].The effectof NKX2-5 on Myocd promoter activity was furthershown to be modulated eitherpositively or negatively through direct interactions between NKX2-5 and eitherCDC7[62]or SMAD3[63],respectively. An upstream NFATc3 binding site was demonstrated in the rat Myocd promoterusing ChIP assaysand forced expression of constitutively active NFATc3 could stimulate both Myocd promoter activity and mRNA expression[64].Interestingly,cyclosporine A,which inhibits NFAT activity,reduces Myocd mRNA and SMC markersboth in vitro and in vivo upon delivery to the vessel wall[65].Another putative activator of Myocd expression is the RNA-binding heterogeneous nuclearribonucleoprotein A1(HNRNPA1)factor,which binds to a proximalregion ofthe mouse Myocd promoterand stimulates both promoteractivity and endogenous mRNA expression in embryonic stem cells[66].Of note,HNRNPA1 could also bind and stimulate the Srf and Mef2c promotersand physically interacted with SRF to enhance SMC contractile gene expression[66].Levels of Myocd mRNA are reduced upon inactivation of SRF in a knockout mouse model[67];however,extensive luciferase assays of conserved CArG elements around the Myocd locus have failed to demonstrate significant SRF-dependent activation(authorˊs unpublished data).Nevertheless,a recent study showed TET2 binding around a CArG box in the human MYOCD promoter with expected demethylation as shown by ChIP assays forthe repressive methylation mark,H3K27me3[68].TET2 was proposed to act upstream of the SRF-MYOCD switch, suggesting that it could be a master orchestrator of the vascular SMC contractile phenotype[68].FOXO3A[69]and KLF4/KLF5[70]bind to regions of the human MYOCD promoter and appearto mediate transcriptionalrepression.Further,embryonic fibroblasts derived from p53 knockoutmice showed elevated Myocd mRNA expression,and wildtype p53 could dose-dependently repress a 1-kb human MYOCD promoter-driven luciferase reporter[71].A short hairpin RNA to p53 blunted TGFβ1-induced Myocd and its targetgenes in SMC[71]. In all of the aforementioned studies,experiments werelimited to in vitro analyses and in mostcases,the conservation of the putative regulatory elementwas either not defined or poorly conserved,indicating need for furthervalidation studies.
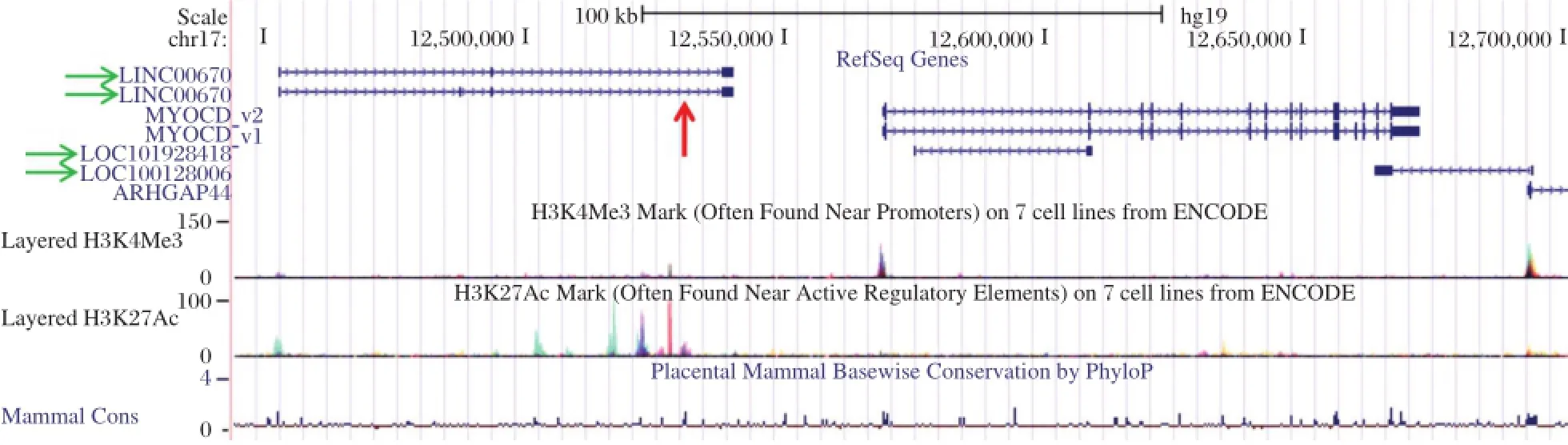
Fig.2Human MYOCD locus.Screenshotof UCSC Genome Browsershowing the two human cardiac muscle MYOCD isoforms(MYOCD_v1 and MYOCD_v2),plus surrounding long noncoding RNA genes(green horizontalarrows)and the approximate location of the human MYOCD enhancer (red verticalarrow).Note the prominent H3K27 acetylation peaks thatmark regulatory elements in close proximity to the enhancer and long noncoding RNA and the smaller H3K4 trimethylation peaks that mark promoters at the 5ˊends of both the MYOCD and ARHGAP44 genes.
Remarkably,only one publication has examined Myocd promoteractivity in the contextofa living animal.Creemers et al.performed an elegant series of reportertiling experiments in mice to show thata distal (~30 kb upstream)enhancer module recapitulated endogenous Myocd expression during embryonic and postnataldevelopment[72].Beta galactosidase reporter gene activity was noted as early as embryonic day 10.0 in the dorsalaorta,consistentwith lineage tracing data(Fig.1).The enhancer region was shown by gel shift,expression screening,and luciferase activities to harbor positively-acting binding sites for MEF2C (whose inactivation results in defective SMC differentiation[73]),FOXO4,and TEAD2[72].No SRF-binding CArG boxes were present in this upstream enhancer region.In this context,MYOCD could activate its own distalenhancerin a MEF2-dependentmannerproviding the firstdemonstration of MYOCD acting in a CArG-SRF-independentfashion[72].Itis also interesting to note thatthe orthologous human distalenhancer lies within an intron of a conserved long intervening noncoding RNA(red arrow in Fig.2).The functionalrelationship of the MYOCD enhancer module to this uncharacterized transcriptis presently unknown.
The study ofthe transcriptionalcontrolof MYOCD is still in its embryonic stages.Several future studies are needed including in vivo validation of key regulatory elements through either BAC recombineering,as shown for the CNN1 gene[74]or through CRISPR/ Cas9-mediated genome editing[75].Second,several long noncoding RNA genes overlap or are in close proximity to MYOCD(green arrows in Fig.2),and these could act in cis to affect MYOCD expression. In this context,a recent study proposed that SENCR, a long noncoding RNA expressed abundantly in vascularendothelialcellsand SMC,contributesto a SMC differentiated state through undefined effects on MYOCD mRNA expression[76].Finally,itwillbe instructive to determine whether any SNPs existin validated regulatory elements thatcould compromise MYOCD expression and the normal SMC contractile phenotype.
Post-transcriptional control of myocardin expression
Essentially allprotein-coding genes undergo alternative splicing and MYOCD is no exception.Initial reports defined two isoforms of MYOCD thatdiffered by inclusion of a 48 amino acid exon 10a(originally referred to as MYOCD-A)[29,61].siRNAknockdown studies targeting this alternate exon suggesta repressor function[77].Of note,ERK1/2 mediated phosphorylation of MYOCD was shown previously to diminish MYOCD transactivation[78],though none of the phosphorylation sitesmap to thisexon.However,a putative ERK1/2 phosphorylation site was found in exon 10a,suggesting that there may be novel functions yet to be defined[79]. Subsequentwork described inclusion ofan exon(exon 2a)containing a premature stop codon resulting in downstream usage of a methionine within exon 4 thatgenerates a SMC-restricted MYOCD isoform[80]. Excluding exon 2a results in a longer isoform of MYOCD thatexhibits a more cardiac muscle-restricted pattern of expression[80].Thus,there are atleast4 isoforms of MYOCD(v1-v4)with MYOCD_v1 and MYOCD_v2 encoding longer,cardiac muscle-restricted isoforms of amino acid lengths 983 and 935(Fig.2) and MYOCD_v3 and MYOCD_v4 encoding shorter, SMC-restricted isoforms of amino acid lengths 904and 856[79].Similar splice variants were described in human tissues[81].
Recently,an RNA-binding protein known as Quaking (QK1)was shown to regulate the balance of MYOCD isoforms in vascular SMC through directinteractions with the pre-mRNA of MYOCD[82].Normalcontractile SMC exhibitlittle expression of QK1 and express the dominant MYOCD_v3 isoform;however,QK1 expression waselevated in vascularlesionsand shown to promote expression ofthe longer,cardiac muscleisoform of MYOCD(MYOCD_v1)[82].Interestingly,the MYOCD_ v3 isoform was more effective in blocking proliferation of SMC and promoting the contractile state than MYOCD_v1 suggesting that a delicate balance of MYOCD isoforms exists,in vascular SMC to maintain normalvesselwall homeostasis[82].Formaldefinition of the function of each MYOCD isoform willrequire precision-guided genome editing using the CRISPR/ Cas9 methodology[75].
Another mechanism for the post-transcriptional controlof MYOCD expression is through microRNA-mediated mRNA deadenylation and degradation through miR-binding sitesin the~5-kb 3ˊun-translated region of MYOCD.The miR-143/145 clusteris a major mediatorofthe SMCcontractilephenotype,in part,through itsdirectorindirecteffecton levelsof MYOCD[83].There is a miR-145 binding site in the 3ˊun-translated region of MYOCD thatsomehow augments its stability and or translation[83].Ofnote,miR-143/145 expression proceeds in a SRF/MYOCD-dependentmanner via an upstream CArG box[83,84]and studies in human coronary artery SMC have shown that TGFβ1,a potent stimulus for the SMC differentiated phenotype[85],signals to induce miR143/145 expression via CArG and adjacent SMAD binding sites[86].Binding sites for miR-145 also exist in the KLF4 and KLF5 transcripts,and evidence exists showing conventionalrepression ofthese known antagonistsof MYOCD expression[83,84].KLF4 isalso targeted by miR-146a[87],miR-25[88],and miR-1[89],leading to an indirectrepressive action on MYOCD expression.Another mechanism of post-transcriptionalcontrolof MYOCD is through miR-221-mediated repression of KIT,which apparently can activate MYOCD gene expression[90].
Recently,a miR-1 binding site was demonstrated in the 3ˊun-translated region of MYOCD,and levels of MYOCD_v3(SMC enriched)were higher in miR-1 nullmice as were several SMC-restricted genes[91,92]. Thus,miR-1 isa negative regulatorofSMCgene expression in the heart.This post-transcriptionalprocess of MYOCD regulation could explain why there isonly transientexpression of many SMC-restricted genes in the embryonic heart,only to re-emerge in adulthearts undergoing failure.There are other miR-binding sites in the MYOCD 3ˊun-translated region including miR-9[64]and miR-135b[93]thateffectchanges in cardiac hypertrophy and cellular growth,respectively.It will be importantto define the dynamic interplay among the known and yet-to-be-defined micro RNAs that bind and regulate MYOCD expression.In particular,itwill be instructive to study the functionality ofthese binding sites in propercontext,such as in a living animal.
Post-translational control of myocardin expression and activity
More than 200 post-translationalmodifications have been defined thatmediate a proteinˊs stability,localization or functional activity,often times through direct protein-protein interactions.A recent review summarized some of the chemicalmodifications of MYOCD protein[94],and a partiallisting ofprotein-protein interactions associated with MYOCD post-translational modifications or attending activity is provided in the Table.As the Table indicates,in mostcases,MYOCD association with anotherprotein alters activity through changes in MYOCD-SRF binding.GSK3B mediates MYOCD phosphorylation at severalserine residues; inhibiting GSK3B accentuated MYOCD-dependent cardiac gene activation and hypertrophy of cultured cardiomyocytes,suggesting that MYOCD phosphorylation may be an important control mechanism for pathophysiological cardiac hypertrophy[95].MYOCD undergoes sumoylation on K445 and this modification appearsto increase itstransactivation overcardiac muscle-specific genes since a K445R mutant exhibits reduced activity[96].MYOCDcan also be ubiquitinylated and this post-translationalchange has been linked to reduced levels of MYOCD via the proteasome[97].On the other hand,UBR5,an ubiquitin E3 ligase,interacts with MYOCD and stabilizes its protein expression, thereby enhancing transactivation of SMC-restricted genes;down-regulation of UBR5 resulted in a decrease in MYOCD-induced SMC gene expression[98].There may be other ubiquitin ligases that interact with MYOCD in certain contexts to enhance or attenuate expression and/or activity.
MYOCD is known to recruitchromatin remodeling factors such as p300 that acetylate lysine residues in histones tails,leading to augmented gene transcription[99].More recently,an elegant study demonstrated p300-dependent acetylation of the MYOCD protein. Acetylation occurs at the amino-terminus and appears to enhance the association of MYOCD with SRF over CArG boxes,thereby increasing SMC target gene expression.Acetylation of MYOCDwas also associated with displacement of HDAC5,a repressor of geneexpression[100].Thus,we see how a factor(p300)serves a dual role of acetylating both localchromatin and the MYOCD cofactor itself to mediate gene expression. Whether these processes are mutually exclusive or facultative remains to be sorted out.Interestingly, ERK1/2 phosphorylation of MYOCD as well as serine-to-aspartic acid phosphomimetics of MYOCD reduce the interaction between MYOCD and p300 showing that different modifications of MYOCD can exhibit opposing activities[78].This implies complex and dynamic,context-dependent changes in MYOCD modifications and subsequent activities necessary to effect moment-to-moment cellular changes in SMC and cardiac muscle cell phenotype.Since MYOCD undergoes phosphorylation,itmustalso interactwith phosphatases thatremove phosphate groups from key amino acid residues;however,no studies to date have formally demonstrated dephosphorylation of MYOCD through a specific phosphatase.There are a numberof other regulatory post-translationalmodifications such as methylation,hydroxylation,oxidation,sulfation, and glycosylation(to name justa few)thathave yetto be demonstrated in MYOCD.Post-translationalmodification ofMYOCD in normaldevelopmentaland pathologicalconditionsisan open area offuture investigation and willbe importantto fully elucidate the biology of this importantcofactorof gene expression.
Diversity in function of myocardin
Although much ofthe literature on MYOCD reports its role as a strong cofactor of SRF directing SMC and cardiacmusclegene expression,there isincreasing recognition ofits biologicalactivities thatare independentof CArG-SRF in these muscle celltypes ordependenton CArG-SRF in a non-muscle celltype.For example,a recentstudy showed that MYOCD interacts with the pancreatic beta cell-specific transcription factor,PDX1, to synergistically activate insulin expression in human mesenchymalstem cells,suggesting thatan improved method may existto generate insulin-expressing beta cells fortype Idiabetes[101].Further,a numberofunanticipated functionsof MYOCDhave emerged such asits role in blocking cellgrowth and inhibiting the skeletal muscle program ofdifferentiation.Thus,like many proteins,MYOCD exhibits a numberofdisparate biologicalactivities and a brief summary ofthese follows.
CArG-independent stimulatory functions of myocardin on muscle gene expression
As discussed above,MYOCD appears to enhance its own expression through association with MEF2 over a MEF2-binding site located in an upstream(CArG-less) enhancer region,presumably through the well-defined MEF2 interaction site located in the amino-terminal RPEL domain of MYOCD[72].MYOCD binds to SMAD3 over a SMAD response elementand increases Tagln(aka Sm22α)promoteractivity independentofthe upstream CArG elements in this promoter[102].MYOCD can interactwith the DNA-binding domain of GATA4 independentof SRF and stimulate cardiac muscle gene expression.Interestingly,the stimulatory effect of MYOCD-GATA4 does notrequire the transactivation domain of GATA4,which when presentacts to repress cardiac muscle genes[103].Another reportshowed that MYOCD can interactwith the TBX5 transcription factorto directcardiac muscle(butnotSMC)gene expression through the TBX-binding site in cardiac muscle gene promoters[104].This study provides some insight into how MYOCD distinguishes between targetgenes associated with cardiac muscle versus SMC gene programs.ITGA8,an integrin alpha subunithighly enriched in SMC,isstimulated by MYOCD independentof SRF ora CArG element[105].Itis unclearas to how MYOCD stimulates this and perhaps other genes that do not otherwise require the CArG-SRF binary complex for gene activation.Further,itwillbe importantto assess the full complementof MYOCD-dependent,CArGSRF-independentgenes in cells where SRF is lacking using Ch IP-seq and RNA-seq following MYOCD overexpression.This will require new antibodies to MYOCD such as one recognizing an endogenous,epitope-tagged MYOCD protein.
Growth inhibitory action of myocardin
Given the dominantrole of MYOCD in establishing a SMC contractile phenotype,itwas perhaps notsurprising to find thatitsoverexpression resulted in reduced proliferation of stably-transfected cells[30].This early finding was corroborated by a series of subsequent studies showing MYOCD-dependentgrowth inhibition in SMC associated with blunted action of NF k B[106], reduced cyclin D1 expression[51],and miR-1 induction[107].Interestingly,there is also evidence forloss in MYOCD expression in certain neoplasms,and the expression of MYOCD has been shown to block cancer cellphenotype through its powerfulorchestration ofthe SMC contractile state[108,109].Future challenges include the elucidation of MYOCD-dependent cyclin D1 repression as well as other genes that are likely to be influenced by the MYOCD cofactor(e.g.,induction of tumor suppressor genes).
Repression of skeletal muscle differentiation
SRF is most abundant in all three muscle types where high level expression of SRF-dependent con-tractile genes exist[110].In addition to SRF,there are some SRF-dependentgenes thatare expressed transiently in each of the three muscle cell types during development(e.g.,Acta2 and Tagln)[111].Importantly, whereas embryonic cardiac muscle and SMC lineages exhibitoverlapping patterns of SMC gene expression, there is no such overlap in gene expression between SMC and skeletalmuscle(Fig.3).The molecular basis forthislack ofoverlap in gene expression between skeletalmuscle and SMC wasnotknown untilthe serendipitous finding that MYOCD completely blocks expression ofmyogenin,a skeletalmuscle masterregulatory gene[31].Repression occurred at the level of the myogenin promoter via MYOCD binding MYOD1, thereby competitively inhibiting MYOD1 binding to an E-box in the myogenin promoter[31].Consistentwith these findings,MYOCD overexpression was shown to inhibit atrogin expression and the skeletal muscle phenotype while knockdown of atrogin elevated MYOCD in the L6 skeletalmuscle cellprecursor[112].The incompatibility of the SMC and skeletalmuscle gene programs is consistent with developmental lineage decisions where common progenitors in the somite give rise to eitherskeletalmuscle orvascularSMC[113].In this context,lineage tracing studies suggestthatMYOCD is transiently expressed within the somite invoking the hypothesis that somitic progenitors with MYOCD expression may be fated forvascular SMC[31,114].
Miscellaneous functions of myocardin
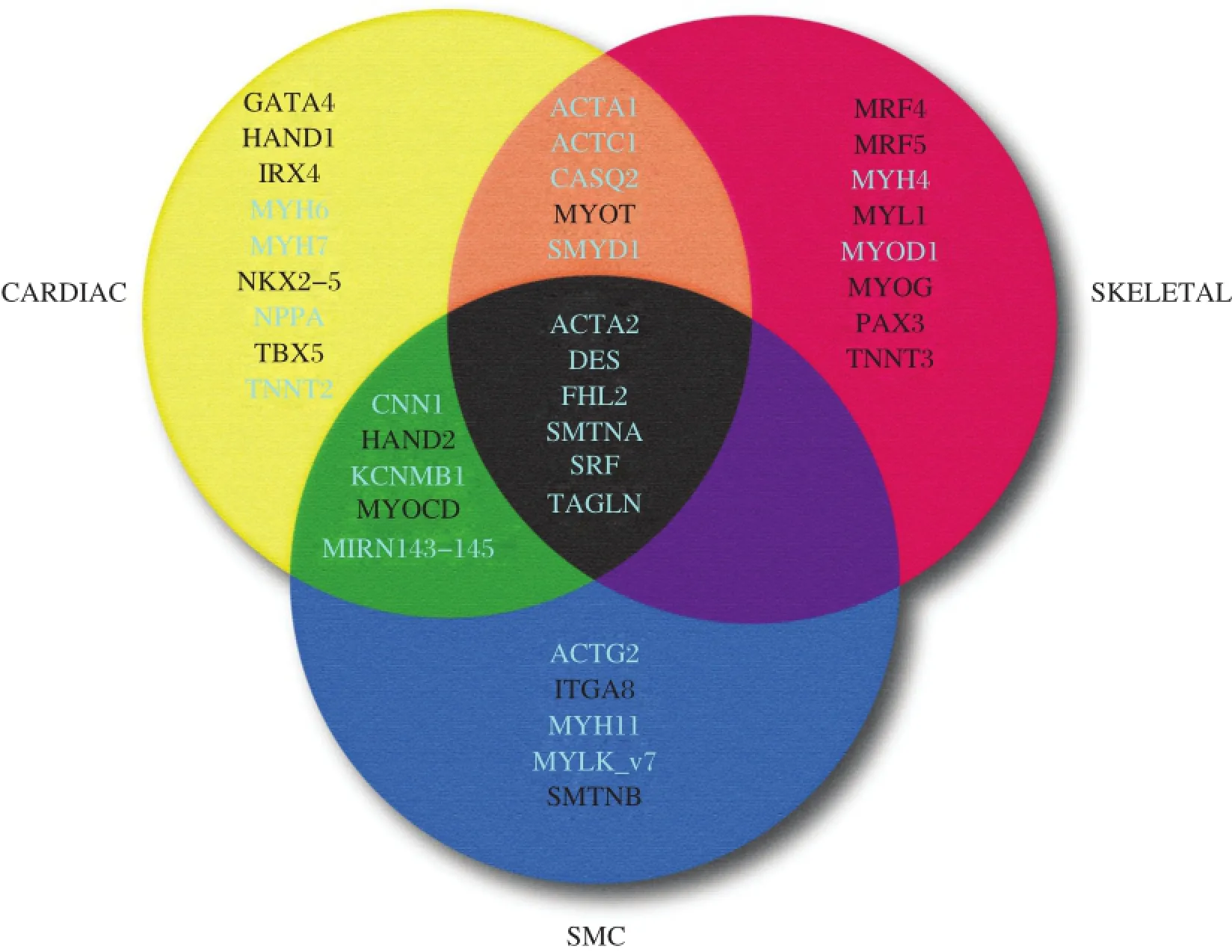
Fig.33-set Venn diagram of genes across the three muscle cell types.Shown are the most specific genes unique to each of the indicated muscle celltypes.White-labeled genes are those that lack functionally-validated CArG boxes while the genes labeled in light color harbor functional CArG boxes.Genes common to the three muscle cell types during development(center)each have atleast one functional CArG box.Whereas several genes are presentatthe intersection of skeletaland cardiac muscle(orange)and cardiac and SMC(green),no genes are found atthe intersection of SMC and skeletal muscle(purple).See the text for discussion.
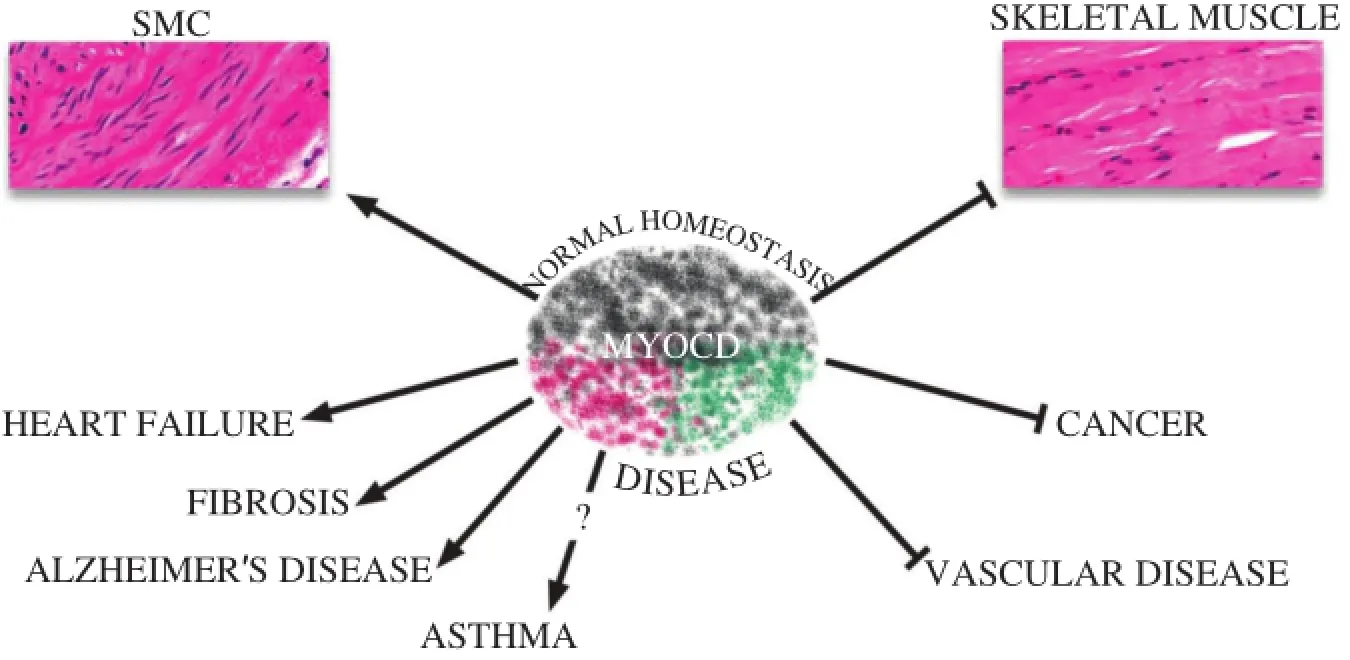
Fig.4Diverse roles of MYOCD in normal homeostasis and disease.Shaded colors of MYOCD icon reflect its diversity in activity,most of which relates to the maintenance of normal homeostasis(SMC differentiation and repression of the skeletal muscle phenotype).MYOCD may contribute to diseases(shade of red)or attenuate diseases(shade of green).See the text for details.
In order for cells to contract,there mustbe careful transcriptionalcontrolofion currents across cellmembranes.Surprisingly,little hasbeen done to link MYOCD to specific activation ofion channelgenes.The KCNMB1 gene,which is highly restricted to SMC,isa directtarget of MYOCD though two CArG-SRF sites in 5ˊuntranslated and proximalintronic regions[48].Genetic prooffor the function ofthese CArG elements was demonstrated in transgenic mouseexperiments[48].Remarkably,ectopic expression of MYOCD can elicition currentactivity in cultured cells[48,53].Interestingly,ion currents,particularly voltage-dependentcalcium entry,can elicit MYOCD expression and various SMC contractile genes[37,115]. Theseintriguing findingsunderscorethe importantconceptof excitation-transcription coupling[116],and highlightthe gap in understanding how channelactivity mediates MYOCD gene transcription or stability[117]. Given the wide number of ion channels expressed in both cardiac muscle and SMC[118,119],itwillbe important to delineate those channels whose expression requires MYOCD and whether their expression proceeds in a CArG-SRF-dependent or CArG-SRF-independent manner.There isalso a growing appreciation fortherole of MYOCD in activation ofmicroRNAs such as miR-145[83]and miR-1[107].Of note,some microRNAs may be repressed by ectopic MYOCD expression,including miR-199a-3p and miR-214[120].That MYOCD may repress gene transcription(miRs,myogenin),emphasizing the need for more study of the binding partners of MYOCD in the nucleus that directly or indirectly influence chromatin architecture as wellas RNA polymerase II-dependent transcription.Finally,as more RNA-seq experiments are undertaken,itwillbe informative to further advance our und erstand ing of MYOCD-dependentgene expression.
Pathological processes linked to myocardin
There is a growing literature documenting changes in MYOCD expression in association with a number of diseases.In addition,MYOCD has been shown to attenuate or exacerbate disease phenotypes,highlighting its potentialas a therapeutic agentortargetfordisease prevention(Fig.4).Much of the literature describes altered expression of MYOCD in diseases of the heartor vasculature,butthere are other disease states(both clinicaland experimental)linked orcorrelated with changes in MYOCD expression.
Cardiovascular diseases
Heart failure is a pervasive clinical problem in Western countries and is a leading cause of morbidity and mortality.There are severalcauses ofheartfailure including ischemic heartdisease and hypertension with the terminalstagescharacterized as dilated cardiomyopathy(DCM).Levels of MYOCD mRNAwere firstshown to be elevated in humans with DCM as compared to normalhearts[81].Moreover,there have been a series of clinicalstudies showing increases in MYOCD mRNA in circulating cells or cardiac tissue from patients with idiopathic cardiomyopathy[121,122],hypertrophic cardiomyopathy[123],and essentialhypertension[124].Interestingly, variants in the 5ˊpromoter region of human MYOCD were associated with reduced levels of circulating MYOCD,which correlated with attenuated leftventricular mass in hypertrophic cardiomyopathy patients[125]. The function of these promoter variants is unknown at this time,butthey presumably interfere with the transcriptionalregulation of MYOCD.A rare missense mutation(K259R)of MYOCD hasbeen reported in acongenital heartdisease patient,and this mutation alters SRF binding to the cardiac,butnotthe SMC,isoformsof MYOCD[126]. Increases in MYOCD transcripts were observed with elevations in GATA4 and NKX2-5 in patients with 3-vesselcoronary artery disease[127].The latterclinicaldata are congruentwith experimentalstudiesshowing forced expression of MYOCD provoking impaired leftventricularsystolic function and electricalactivity in the piglet[128]as well as hypertrophy of cardiomyocytes[122]. Importantly,silencing MYOCD via intramyocardial delivery ofa shorthairpin RNA in a doxorubicin-induced modelofheartfailure,normalized increases in MYOCD and severalfetalgenes(including various SMC genes)and attenuated cardiac muscle dysfunction and death[129]. Collectively,these clinical and experimental data suggestthat MYOCD represents a viable therapeutic target/biomarker in the setting of cardiac hypertrophy and failure.
Whereas MYOCD levels increase in association with cardiac disease,a number of reports have found MYOCD mRNA to be transiently down-regulated following acute injuries to the blood vesselwallin differen t animal models[130-134].A recent elegant study showed thatMYOCDdelivery to the vesselwallblocked the neointimal response to vascular injury through microRNA-mediated suppression ofthe Pdgfrb gene[132]. This resultsuggests therapeutic potentialfor enhanced MYOCD expression in the contextofacute injuriessuch as those encountered in the clinic where there remains some residual restenosis rates following balloon angioplasty ofatherosclerotic lesions.MYOCD levels are also reduced in experimentalmodels of atherosclerosis[135]and hypertension[136].Itremains to be studied whether ectopic MYOCD exerts any beneficialeffectin these chronic conditions of vascular disease.MYOCD mRNA levels were reported to be reduced in venular SMC of experimentally-induced and clinically-diagnosed variocose veins[137].Interestingly,treatmentwith Bortezomib, (a proteasome inhibitor)enhanced MYOCD expression and reduced the proliferation and migration of SMC as wellas the varicosity ofveins in a mouse model[137].
Pulmonary hypertension is associated with hypoxia and the formation ofso-called plexiform lesions.Hypoxia induces Midkine which has been linked to increases in MYOCD in the setting of pulmonary hypertension[138], and these findingsare congruentwith hypoxia-mediated increases in MYOCD in other models of pulmonary hypertension[139,140].Hypoxia has also been shown to induce MYOCD in human cerebral SMC as well as rodent SMC[141].Hypoxia-induced MYOCD in this setting appearsto be linked to faulty amyloid beta clearance and the emergence of cerebral amyloid angiopathy, which is pathognomonic of Alzheimerˊs disease[141].Of note,a putative hypoxia response element was found in the 5ˊpromoter region of Myocardin[141];however, formal proof that this elementbinds to HIF1A and underlies the induction of MYOCD by hypoxia awaits further study.Interestingly,increases in MYOCD and the SMC contractile program have also been observed in Alzheimerˊs disease patient-derived cerebral SMC with consequent hyper-contractility and hypoperfusion, which are additionalfeatures of Alzheimerˊs disease in humans[142].Thus,Alzheimerˊs disease and attending neurodegeneration and cognitive decline may result from elevated MYOCD expression with poor amyloid clearance and hypoperfusion due to hypercontractile vascular SMC[143].This would suggesta more vascularcentric(as opposed to neuro-centric)view for the pathogenesis of this devastating neurologicaldisorder. Although the weightofevidence supports hypoxia as a stimulus for MYOCD induction,there are studies that show a negative correlation between MYOCD levels and hypoxia,highlighting the context-dependentnature of these responses[144,145].Finally,there is one report of increased MYOCD in calcified aortic valve cusps with accompanying elevations in SMC markers,suggesting that SMC-derived MYOCD could play a role in the pathogenesis ofcalcific aortic stenosis[146].There remain other vascular disorders where altered MYOCD may be of functional consequence,including peripheral artery disease,aneurysm formation,and transplant arteriopathy.The integration of inducible isoforms of MYOCD in animalgenomes using CRISPR-Cas9 technology willbe of major interestto further assess the efficacy of MYOCD in thwarting a number of vascular disorders.
Cancer
The consistent finding that MYOCD suppresses normalcellgrowth is consonantwith the increasing evidence supporting a tumor suppressor role for this SRF cofactor in the setting of cancer[147].One clinicalstudy has shown attenuated MYOCD in severalnasopharyngealcarcinoma celllines,and the decrease in MYOCD correlated with hypermethylation of its promoter[148]. Notably,treatment of these cancer cells with the demethylating agent,5-azacytidine,increased MYOCD expression,further suggesting the tumor suppressive nature of MYOCD[148].Sometimes,the tumor suppressor effectof MYOCD may be indirect.Forexample,Maspin is transcriptionally induced by MYOCD and the upregulation of Maspin leads to apoptosis of breastcancer cells[149].In uterine leiomyosarcoma celllines,MYOCD shows clear growth suppressive properties,in part through the activation of the p21 growth inhibitor via SRF-binding CArG element[109],butalso because of the induction of several contractile genes such as CNN1,which itself has been labeled a tumor suppressor gene[150].In thiscontext,CNN1 and other SMC contractile genes have been demonstrated to be reduced in metastatic human tumors and form part of a molecular signature for the metastatic phenotype[151].Whether reductionsin these genes,allofwhich carry SRF-binding CArG boxes,stem from reduced levels of MYOCD ispresently unknown.Itmustbe stressed,however,that there are some instances where levels of MYOCD are elevated in tumor cells[152],further underscoring thecomplexity of human disease and the need to exercise circumspection when making broad assumptions over the importance of this or any other protein in human pathology.On one finalnote,itis intriguing to consider the fact that while vascular SMC exhibit phenotypic plasticity,vascular leiomyosarcomas are exceedingly rare cancers[153].The molecular basis for such scarce tumors is completely unknown,but may well relate to the SMC-restricted isoforms of MYOCD that appear to exertgreater growth inhibitory action than the longer, cardiac muscle-enriched isoforms[91].
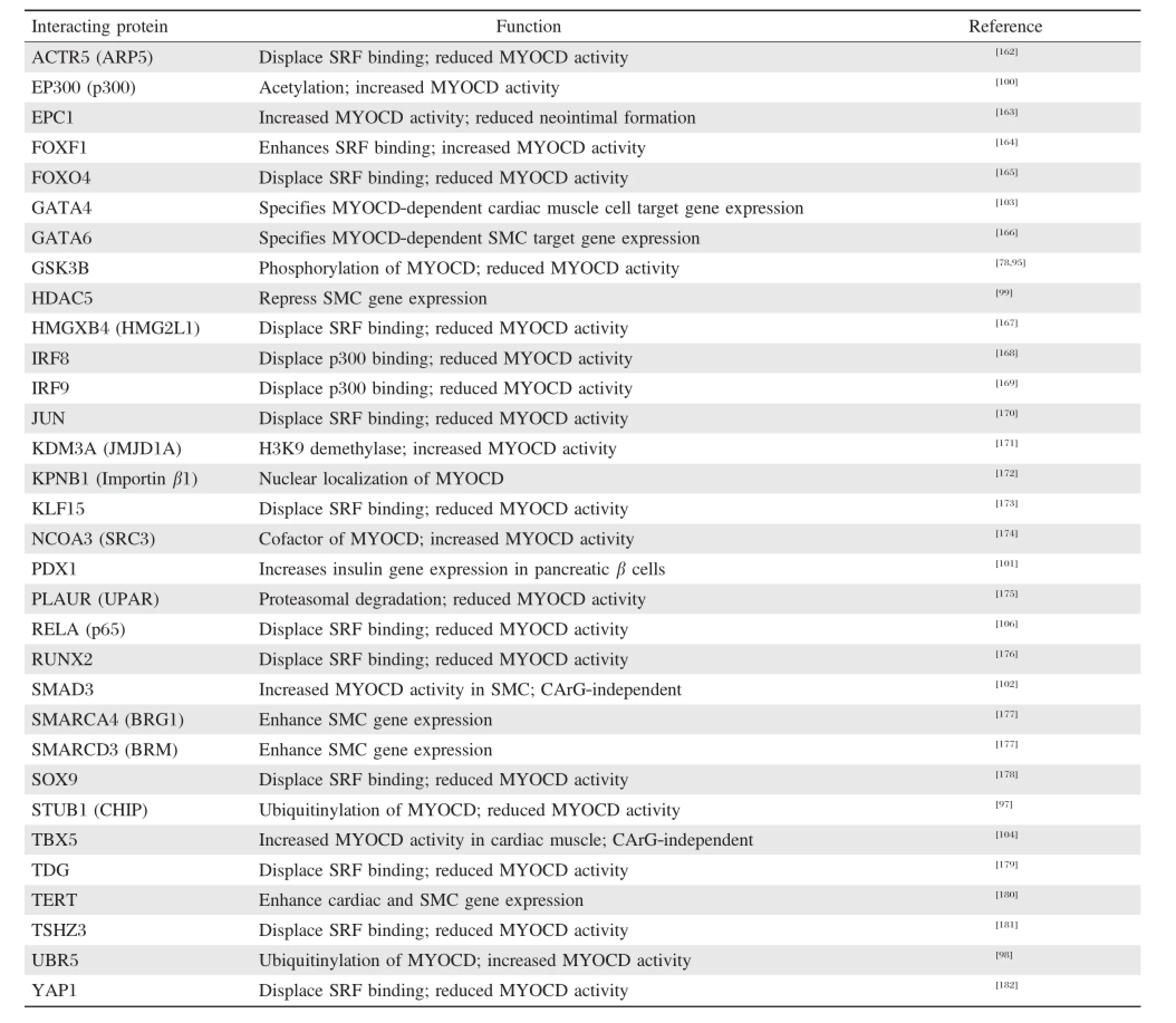
Table 1 Myocardin(MYOCD)-interacting proteins and functional consequence
Diabetes
Type 2 diabetes has ascended to epidemic proportions and will increasingly strain Western health care systems.One of the manifestations of type 2 diabetes is a spike in RAGE with inflammation and calcification.Overexpression of RAGE suppresses MYOCD and SMC contractile genes and favors an osteogenic phenotype,which can lead to calcification of arteries[154].In addition to vascular complications,diabetic patients often present with erectile dysfunction. Experimental models of diabetes have consistently shown a reduced expression of MYOCD in the corpus cavernosum[155,156].From a therapeutic standpoint, ectopic MYOCD was shown to reconstitute normal erectile function in diabetic rats through the conversion of proliferating SMC within the corpus cavernosum to a contractile state[157].Other complications of diabetes include accelerated atherosclerosis,retinalangiopathy, and peripheral artery disease all of which have yetto be examined in the context of MYOCD.
Miscellaneous diseases
There are several other diseases in which MYOCD has been studied.For example,in a modelofintestinal obstruction,MYOCD mRNA levels were shown to be reduced concomitantwith several MYOCD-dependent SMC contractile genes[158].In one ofthe firstexamplesof Myocd haploinsufficiency,micewith only one functional Myocd allele,exhibited bladder SMC hypersensitivity. This was surmised to resultfrom the reduced expression ofmiR-1 and a corresponding increase in a known miR-1 targetmRNA,Gja1 orconnexin 43.Adultmice with one copy of Myocd showed lower bladdercapacity consistentwith a hypersensitive phenotype[159].Hepatic stellate cells can undergo transdifferentiation to a myofibroblastlike phenotype in liver diseases associated with fibrosis, and studies have shown MYOCD is induced in experimental models of liver fibrosis[160,161].Interestingly, reducing MYOCD with siRNA predictably normalized the myofibroblastphenotype,suggesting that MYOCD (and probably itsrelated MRTFs)could bea noveltarget forthe treatmentofliver fibrosis[161].
Future perspectives
The last 10 years of research on MYOCD have focused mainly on its wellaccepted role as the principal component to a switch for the SMC differentiated state. There have been new insights into MYOCD regulation, its association with otherproteins,as wellas its expression and potentialutility as a markerortargetoftherapy forseveraldiseases.Work in the nextdecade should be focused on thedevelopmentofnew reagents(antibodies) and animalmodels(inducible expression of MYOCD)to furtherunderstand this remarkable cofactorˊs function in normaland pathologicalprocesses.In addition,there should be effortdevoted towards fully elucidating transcriptomes under controlof MYOCD,particularly the expanding class oflong non-coding RNAs.Further,we need to define the mechanisms through which MYOCD functions independently of SRF.What other DNA binding factors does MYOCD interactwith and in what contexts?There are SMC-associated diseases in which virtually nothing is known regarding expression and functionality of MYOCD(e.g.,asthma).Finally,as more and more human genomes are sequenced,itwill be informative to define the functionality of sequence variantssuch as SNPsboth in and around MYOCD coding and non-coding sequence space.
[1]Davis RL,Weintraub H,Lassar AB.Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987;51(6):987-1000.
[2]Olson EN.MyoD family:a paradigm for development. Genes Dev 1990;4(9):1454-1461.
[3]Lee JE,Hollenberg SM,Snider L,et al.Conversion of Xenopus ectoderm into neurons by Neuro D,a basic helix-loop-helix protein.Science 1995;268(5212):836-844.
[4]Komuro I,Izumo S.Csx:A murine homeobox-containing gene specifically expressed in the developing heart.Proc Natl Acad Sci U S A 1993;90(17):8145-8149.
[5]Graf T,Enver T.Forcing cells to change lineages.Nature 2009;462(7273):587-594.
[6]Graf T.Historical origins of transdifferentiation and reprogramming.Cell Stem Cell 2011;9(6):504-16.
[7]Majesky MW.Developmental basis of vascular smooth muscle diversity.Arterioscler Thromb Vasc Biol 2007; 27(6):1248-1258.
[8]Campbell GR,Campbell JH.Smooth muscle phenotypic changes in arterial wall homeostasis:Implications for the pathogenesis of atherosclerosis.Exp Mol Pathol 1985;42(2):139-162.
[9]Halayko AJ,Solway J.Molecular mechanisms of phenotypic plasticity in smooth muscle cells.J Appl Physiol 2001;90(1):358-368.
[10]Owens GK,Kumar MS,Wamhoff BR.Molecular regulation ofvascularsmooth muscle celldifferentiation in development and disease.Physiol Rev 2004;84(3):767-801.
[11]Nguyen AT,Gomez D,Bell RD,etal.Smooth muscle cell plasticity:factor fiction?Circ Res 2013;112(1):17-22.
[12]Patel CV,Gorski DH,LePage DF,etal.Molecular cloning o f a ho meobox tran scription factor from adult aortic smooth muscle.J Biol Chem 1992;267(36):26085-26090.
[13]Miano JM,Firulli AB,Olson EN,etal.Restricted expression of homeobox genes distinguishes fetal from adult human smooth muscle cells.Proc Natl Acad Sci U S A 1996;93(2):900-905.
[14]Hsieh CM,Yoshizumi M,Endege WO,et al.APEG-1,a novelgene preferentially expressed in aortic smooth muscle cells,is down-regulated by vascular injury.J Biol Chem 1996;271(29):17354-17359.
[15]Watanabe M,Layne MD,Hsieh CM,etal.Regulation of smooth muscle cell differentiation by AT-rich interaction domain transcription factors Mrf2alpha and Mrf2beta. Circ Res 2002;91(5):382-389.
[16]Layne MD,Endege WO,Jain MK,et al.Aortic carboxypeptidase-like protein,a novel protein with discoidin and carboxypeptidase-like domains,is up-regulated during vascular smooth muscle cell differentiation.J Biol Chem 1998;273(25):15654-15660.
[17]Chin MT,Maemura K,Fukumoto S,etal.Cardiovascular basic helix loop helix factor 1,a novel transcriptional repressor expressed preferentially in the developing and adult cardiovascular system.J Biol Chem 2000;275(9): 6381-6387.
[18]Kawai-Kowase K,Kumar MS,Hoofnagle MH,et al. PIAS1 activates the expression ofsmooth muscle celldifferentiation marker genes by interacting with serum response factor and class Ibasic helix-loop-helix proteins. Mol Cell Biol 2005;25(18):8009-8023.
[19]Wang DZ,Chang PS,Wang Z,etal.Activation ofcardiac gene expression by myocardin,a transcriptional cofactor for serum response factor.Cell 2001;105(7):851-862.
[20]Yamada M,Tsuji S,Takahashi H.Pathology of CAG repeat diseases.Neuropathology 2000;20(4):319-325.
[21]Wang Z,Wang DZ,Pipes GCT,et al.Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci U S A 2003;100(12):7129-7134.
[22]Minty A,Kedes L.Upstream regions of the human cardiac actin gene that modulate its transcription in muscle cells:presence of an evolutionarily conserved repeated motif.Mol Cell Biol 1986;6(6):2125-2136.
[23]Miano JM.Serum response factor:toggling between disparate programs of gene expression.J Mol Cell Cardiol 2003;35(6):577-593.
[24]Norman C,Runswick M,Pollock R,et al.Isolation and properties of cDNA clones encoding SRF,a transcription factor thatbinds to the c-fos serum response element.Cell 1988;55(6):989-1003.
[25]Cen B,Selvaraj A,Prywes R.Myocardin/MKL family of SRF coactivators:key regulators of immediate early and muscle specific gene expression.J Cell Biochem 2004; 93(1):74-82.
[26]Wang DZ,Olson EN.Controlofsmooth muscle developmentby the myocardin family oftranscriptionalcoactivators.Curr Opin Genet Dev 2004;14(5):558-566.
[27]Pipes GCT,Creemers EE,Olson EN.The myocardin family of transcriptional coactivators:versatile regulators of cell growth,migration,and myogenesis.Genes Dev 2006;20(12):1545-1556.
[28]Parmacek MS.Myocardin-related transcription factors: critical coactivators regulating cardiovascular development and adaptation.Circ Res 2007;100(5):633-644.
[29]Du K,Ip HS,Li J,et al.Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation.Mol Cell Biol 2003;23(7):2425-2437.
[30]Chen J,Kitchen CM,Streb JW,etal.Myocardin:a componentof a molecular switch for smooth muscle differentiation.J Mol Cell Cardiol 2002;34(10):1345-1356.
[31]Long X,Creemers EE,Wang DZ,et al.Myocardin is a bifunctionalswitch forsmooth versus skeletalmuscle differentiation.Proc NatlAcad SciU S A 2007;104(42):16570-16575.
[32]Small EM,Warkman AS,Wang DZ,et al.Myocardin is sufficient and necessary for cardiac gene expression in Xenopus.Development 2005;132:987-997.
[33]Wark man AS,Yatsk ievych TA,Hardy KM,et al. Myocardin expression during avian embryonic heart development requires the endoderm but is independent of BMP signaling.Dev Dyn 2008;237(1):216-221.
[34]Yoshida T,Sinha S,Dandre F,et al.Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes.Circ Res 2003;92(8):856-864.
[35]Yoshid a T,Kawai-Kowase K,Owens GK.Forced expression of myocardin is not sufficient for induction of smooth muscle differentiation in multipotential cells. Arterioscler Thromb Vasc Biol 2004;24(9):1596-1601.
[36]Miano JM,Berk BC.Retinoids:Versatile biological response modifiers of vascular smooth muscle phenotype. Circ Res 2000;87(5):355-362.
[37]Wamhoff BR,Bowles DK,McDonald OG,et al.L-type voltage-gated Ca 2+channels modulate expression of smooth muscle differentiation marker genes via a Rho kinase/myocardin/SRF-dependent mechanism.Circ Res 2004;95(4):406-414.
[38]Chiu CZ,Wang BW,Shyu KG.Effects ofcyclic stretch on the molecularregulation ofmyocardin in rataortic vascular smooth muscle cells.J Biomed Sci2013;20(1):50.
[39]Martin K,Weiss S,Metharom P,et al.Thrombin stimulates smooth muscle cell differentiation from peripheral blood mononuclear cells via protease-activated receptor-1,RhoA,and myocardin.Circ Res 2009;105(3):214-218.
[40]Xiao Q,Luo Z,Pepe AE,et al.Embryonic stem cell differentiation into smooth muscle cells is mediated by Nox4-produced H2O2.Am.J.Physiol Cell Physiol 2009; 296(4):C711-C723.
[41]Pepe AE,Xiao Q,ZampetakiA,etal.Crucialrole of Nrf3 in smooth muscle celldifferentiation from stem cells.Circ Res 2010;106(5):870-879.
[42]Kurpinski K,Lam H,Chu J,et al.Transforming growth factor-beta and notch signaling mediate stem cell differentiation into smooth muscle cells.Stem Cells 2010; 28(4):734-742.
[43]Fritz KE,Jarmolych J,Daoud AS.Association of DNA synthesis and apparent dedifferentiation of aortic smooth muscle cells in vitro Exp Mol Pathol1970;12(3):354-362.
[44]Chamley-Campbell J,Campbell GR,Ross R.The smooth muscle cell in culture.Physiol Rev 1979;59(1):1-61.
[45]Iyemere VP,Proudfoot D,Weissberg PL,et al.Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification.J Intern Med 2006;260(3): 192-210.
[46]Alexander MR,Owens GK.Epigenetic controlof smooth muscle cell differentiation and phenotypic switching in vascular development and disease.Annu Rev Physiol 2012;74:13-40.
[47]Miano JM,Kitchen CM,Chen JY,et al.Myocardin is expressed in adult aorta and activates a smooth muscle cell differentiation program.Arterioscler Thromb Vasc Biol 2002;22(5):A2-A2.
[48]Long X,Tharp DL,Georger MA,etal.The smooth muscle cell-restricted KCNMB1 ion channel subunit is a direct transcriptional target of serum response factor and myocardin.J Biol Chem 2009;284(48):33671-33682.
[49]Nanda V,Miano JM.Leiomodin 1:A n ew serum response factor-dependent target gene expressed preferentially in differentiated smooth muscle cells.J Biol Chem 2012;287(4):2459-2467.
[50]Zhou J,Herring BP.Mechanisms responsible for the promoter-specific effects of myocardin.J Bio l Ch em 2005;280(11):10861-10869.
[51]Long X,BellRD,Gerthoffer WT,etal.Myocardin is sufficient for a SMC-like contractile phenotype.Arterioscler Thromb Vasc Biol 2008;28(8):1505-1510.
[52]Jiang Y,Yin H,Zheng XL.MicroRNA-1 inhibits myocardin-induced contractility of human vascular smooth muscle cells.J Cell Physiol 2010;225(2):506-511.
[53]Raphel L,Talasila A,Cheung C,etal.Myocardin overexpression is sufficient for promoting the development of a mature smooth muscle cell-like phenotype from human embryonic stem cells.PLoS One 2012;7(8):e44052.
[54]McDonald OG,Wamhoff BR,Hoofnagle MH,et al. Controlof SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo.J Clin Invest 2006;116(1):36-48.
[55]Ieda M,Fu JD,Delgado-Olguin P,et al.Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors.Cell 2010;142(3):375-386.
[56]Li S,Wang DZ,Richardson JA,etal.The serum response factor coactivator myocardin is required for vascular smooth muscle development.Proc Natl Acad Sci U S A 2003;100(16):9366-9370.
[57]Huang J,Lu MM,Cheng L,et al.Myocardin is required for cardiomyocyte survival and maintenance of heart function.Proc Natl Acad Sci U S A 2009;106(44): 18734-18739.
[58]Huang J,Elicker J,Bowens N,et al.Myocardin regulates BMP10 expression and is required for heartdevelopment. J Clin Invest 2012;122(10):3678-91.
[59]Hoofnagle MH,Neppl RL,Berzin EL,etal.Myocardin is differentially required for the development of smooth muscle cells and cardiomyocytes.Am J Physiol Heart Circ Physiol 2011;300(5):H1707-H1721.
[60]Huang J,Cheng L,Li J,et al.Myocardin regulates expression of contractile genes in smooth muscle cells and is required forclosure ofthe ductus arteriosus in mice. J Clin Invest 2008;118(2):515-525.
[61]Ueyama T,Kasahara H,Ishiwata T,et al.Myocardin expression is regulated by Nkx2.5,and its function is required for cardiomyogenesis.Mol Cell Biol 2003; 23(24):9222-9232.
[62]Shi N,Chen SY.Cell division cycle 7 mediates transforming growth factor-beta-induced smooth muscle maturation through activation of myocardin gene transcription.J Biol Chem 2013;288(48):34336-42.
[63]Xie WB,Li Z,Miano JM,et al.Smad3-mediated myocardin silencing:a novelmechanism governing the initiation of smooth muscle differentiation.J Biol Chem 2011;286(17):15050-15057.
[64]Wang K,Long B,Zhou J,et al.miR-9 and NFATc3 regulate myocardin in cardiac hypertrophy.J Biol Chem 2010;285(16):11903-11912.
[65]Garvey SM,Sinden DS,Schoppee Bortz PD,et al. Cyclosporine up-regulates Kruppel-like factor-4(KLF4) in vascular smooth muscle cells and drives phenotypic modulation in vivo.J Pharmacol Exp Ther 2010;333(1): 34-42.
[66]Huang Y,Lin L,Yu X,etal.Functional involvements of heterogeneous nuclear ribonucleoprotein A1 in smooth muscle differentiation from stem cells in vitro and in vivo. Stem Cells 2013;31(5):906-17.
[67]Miano JM,Ramanan N,Georger MA,et al.Restricted inactivation ofserum response factor to the cardiovascular system.Proc Natl Acad Sci U S A 2004;101(49):17132-17137.
[68]Liu R,Jin Y,Tang WH,et al.Ten-eleven translocation-2 (TET2)is a master regulator of smooth muscle cell plasticity.Circulation 2013;128(18):2047-57.
[69]Yang X,Gong Y,Tang Y,et al.Spry1 and Spry4 differentially regulate human aortic smooth muscle cellphenotype via Akt/Fox O/myocardin signaling.PLo S One 2013;8(3):e58746.
[70]Turner EC,Huang CL,Govindarajan K,etal.Identification of a Klf4-dependent upstream repressor region mediating transcriptionalregulation of the myocardin gene in human smooth muscle cells.Biochim Biophys Acta 2013;1829(11): 1191-201.
[71]Molchadsky A,Shats I,Goldfinger N,et al.p53 plays a role in mesenchymal differentiation programs,in a cell fate dependent manner.PLoS One 2008;3(11):e3707.
[72]Creemers EE,Sutherland LB,McNally J,etal.Myocardin is a direct transcriptional target of Mef2,Tead and Foxo proteins during cardiovascular development.Development 2006;133(21):4245-4256.
[73]Lin Q,Lu J,Yanagisawa H,et al.Requirement of the MADS-box transcription factor MEF2C for vascular development.Development 1998;125(22):4565-4574.
[74]Long X,Slivano OJ,Cowan SL,et al.Smooth muscle calponin:an unconventional CArG-dependent gene that antagonizes neointimal formation.Arterioscler Thromb Vasc Biol 2011;31(10):2172-2180.
[75]Wang H,Yang H,Shivalila CS,etal.One-step generation of mice carrying mutations in multiple genes by CRISPR/ Cas-mediated genome engineering.Cell 2013;153(4): 910-8.
[76]Bell RD,Long X,Lin M,et al.Identification and initial functional characterization of a human vascular cellenriched long noncoding RNA.Arterioscler Thromb Vasc Biol 2014;34(6):1249-59.
[77]Ilagan RM,Genheimer CW,Quinlan SF,et al.Smooth muscle phenotypic diversity is mediated through alterations in myocardin gene splicing.J Cell Physiol 2011; 226(10):2702-2711.
[78]Taurin S,Sandbo N,Yau DM,et al.Phosphorylation of myocardin by extracellular signal regulated protein kinase.J Biol Chem 2009;284(49):33789-33794.
[79]Imamura M,Long X,Nanda V,et al.Expression and functional activity of four myocardin isoforms.Gene 2010;464(1-2):1-10.
[80]Creemers EE,Sutherland LB,Oh J,etal.Coactivation of MEF2 by the SAP domain proteins myocardin and MASTR.Mol Cell 2006;23(1):83-96.
[81]Torrado M,Lc/pez E,Centeno A,etal.Myocardin mRNA is augmented in the failing myocardium:expression profiling in the porcine modeland human dilated cardiomyopathy.J Mol Med 2003;81(9):566-577.
[82]van der Veer EP,de Bruin RG,Kraaijeveld AO,et al. Quaking,an RNA-binding protein,is a critical regulator of vascular smooth muscle cell phenotype.Circ Res 2013;113(9):1065-75.
[83]Cordes KR,Sheehy NT,White MP,et al.miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009;460(7256):705-710.
[84]Xin M,Small EM,Sutherland LB,et al.MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev 2009;23(18):2166-2178.
[85]Tang Y,Yang X,Friesel RE,et al.Mechanisms of TGF-beta-induced differentiation in human vascular smooth muscle cells.J Vasc Res 2011;48(6):485-494.
[86]Long X,Miano JM.Transforming growth factor-b1 (TGF-b1)utilizes distinctpathways for the transcriptional activation of micro RNA 143/145 in human coronary artery smooth muscle cells.J Biol Chem 2011;286(34): 30119-30129.
[87]Sun SG,Zheng B,Han M,et al.miR-146a and Kruppellike factor 4 form a feedback loop to participate in vascular smooth muscle cell proliferation.EMBO Reports 2011;12(1):56-62.
[88]Kuhn AR,Schlauch K,Lao R,et al.MicroRNA expression in human airway smooth muscle cells:role of miR-25 in regulation of airway smooth muscle phenotype. Am J Respir Cell Mol Biol 2010;42(4):506-13.
[89]Xie C,Huang H,Sun X,et al.MicroRNA-1 regulates smooth muscle celldifferentiation by repressing Kruppellike factor 4.Stem Cells Dev 2011;20(2):205-210.
[90]Davis BN,Hilyard AC,Nguyen PH,et al.Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype.J Biol Chem 2009;284(6):3728-3738.
[91]Heidersbach A,Saxby C,Carver-Moore K,etal.microRNA-1 regulates sarcomere formation and suppresses smooth muscle gene expression in the mammalian heart.Elife 2013;2:e01323.
[92]Wystub K,Besser J,Bachmann A,et al.miR-1/133a clusters cooperatively specify the cardiomyogenic lineage by adjustment of myocardin levels during embryonic heart development.PLoS Genet 2013;9(9):e1003793.
[93]Xu WG,Shang YL,Cong XR,et al.Micro RNA-135b promotes proliferation,invasion and migration of osteosarcoma cells by degrading myocardin.Int J Oncol 2014;45(5):2024-32.
[94]Zheng XL.Myocardin and smooth muscle differentiation. Arch Biochem Biophys 2014;543:48-56.
[95]Badorff C,Seeger FH,Zeiher AM,et al.Glycogen synthase kinase 3a inhibits myocardin-dependent transcription and hypertrophy induction through site-specific phosphorylation.Circ Res 2005;97(7):645-654.
[96]Wang J,Li AK,Wang ZG,etal.Myocardin sumoylation transactivates cardiogenic genes in pluripotent 10T1/2 fibroblasts.Mol Cell Biol 2007;27(2):622-632.
[97]Xie P,Fan Y,Zhang H,et al.CHIP represses myocardininduced smooth muscle cell differentiation via ubiquitinmediated proteasomal degradation.Mol Cell Biol 2009;29(9):2398-2408.
[98]Hu G,Wang X,Saunders DN,etal.Modulation of myocardin function by the ubiquitin E3 ligase UBR5.J Biol Chem 2010;285(16):11800-11809.
[99]Cao D,Wang Z,Zhang CL,et al.Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin.Mol Cell Biol 2005;25(1):364-376.
[100]Cao D,Wang C,Tang R,etal.Acetylation of myocardin is required for the activation of cardiac and smooth muscle genes.J Biol Chem 2012;287(46):38495-504.
[101]Li JT,Sun FX.Myocardin and pdx-1 synergistically induce hMSCs to differentiate into insulin secreting cells. Biochem Biophys Res Commun 2014.
[102]Qiu P,Ritchie RP,Fu Z,et al.Myocardin enhances Smad3-mediated transforming growth factor-b1 signaling in a CArG box-independent manner:Smad-binding elementis an important cis element for SM22a transcription in vivo.Circ Res 2005;97(10):983-991.
[103]Oh J,Wang Z,Wang DZ,et al.Target gene-specific modulation of myocardin activity by GATA transcription factors.Mol Cell Biol 2004;24(19):8519-8528.
[104]Wang C,Cao D,Wang Q,et al.Synergistic activation of cardiac genes by myocardin and Tbx5.PLoS One 2011; 6(8):e24242.
[105]Kitchen CM,Cowan SL,Long X,et al.Expression and promoter analysis of a highly restricted integrin alpha gene in vascular smooth muscle.Gene 2013;513(1): 82-89.
[106]Tang Rh,Zheng XL,Callis TE,et al.Myocardin inhibits cellular proliferation by inhibiting NF-k B(p65)-dependent cell cycle progression.Proc Natl Acad Sci U S A 2008;105(9):3362-3367.
[107]Chen J,Yin H,Jiang Y,et al.Induction of microRNA-1 by myocardin in smooth muscle cells inhibits cell proliferation.Arterioscler Thromb Vasc Biol 2011;31(2):368-375.
[108]Milyavsky M,Shats I,Cholostoy A,et al.Inactivation of myocardin and p16 during malignanttransformation contributes to a differentiation defect.Cancer Cell 2007; 11(2):133-146.
[109]Kimura Y,Morita T,Hayashi K,et al.Myocardin functions as an effective inducer of growth arrest and differen tiation in hu man u terine leiomyosarcoma cells. Cancer Res 2010;70(2):501-511.
[110]Belaguli NS,Schildmeyer LA,Schwartz RJ.Organization and myogenic restricted expression of the murine serum response factor gene:a role for autoregulation.J Biol Chem 1997;272(29):18222-18231.
[111]LiL,Miano JM,CserjesiP,etal.SM22a,a marker ofadult smooth muscle,is expressed in multiple myogenic lineages during embryogenesis.Circ Res 1996;78(2):188-195.
[112]Jiang Y,Singh P,Yin H,etal.Opposite roles of myocardin and atrogin-1 in L6 myoblast differentiation.J Cell Physiol 2013;228(10):1989-95.
[113]Lagha M,Brunelli S,Messina G,etal.Pax3:Foxc2 reciprocalrepression in the somite modulates muscular versus vascular cell fate choice in multipotent progenitors.Dev Cell 2009;17(6):892-899.
[114]Espinoza-Lewis RA,Wang DZ.Generation ofa Cre knockin into the Myocardin locus to mark early cardiac and smooth muscle celllineages.Genesis 2014;52(10):879-87.
[115]Ren J,Albinsson S,Hellstrand P.Distinct effects of voltage-and store-dependent calcium influx on stretchinduced differentiation and growth in vascular smooth muscle.J Biol Chem 2010;285(41):31829-31839.
[116]Wamhoff BR,Bowles DK,Owens GK.Excitation-transcription coupling in arterial smooth muscle.Circ Res 2006;98(7):868-878.
[117]Miano JM.Channeling to myocardin.Circ Res 2004; 95:340-342.
[118]Demolomb e S,Marion neau C,Le Bouter S,et al. Functional genomics of cardiac ion channel genes. Cardiovasc Res 2005;67(3):438-47.
[119]Cidad P,Moren o-Dominguez A,Novensa L,et al. Characterization of ion channels involved in the proliferative response of femoral artery smooth muscle cells. Arterioscler Thromb Vasc Biol 2010;30(6):1203-1211.
[120]Park C,Hennig GW,Sanders KM,et al.Serum response factor-dependent micro RNAs regulate gastrointestinal smooth muscle cell phenotypes.Gastroenterology 2011; 141(1):164-175.
[121]KontarakiJE,Parthenakis FI,NyktariEG,etal.Myocardial gene expression alterations in peripheral blood mononuclear cells of patients with idiopathic dilated cardiomyopathy.Eur J Heart Fail2010;12(6):541-8.
[122]Xing W,Zhang TC,Cao D,etal.Myocardin induces cardiomyocyte hypertrophy.Circ Res 2006;98(8):1089-1097.
[123]Kontaraki JE,Parthenakis FI,Patrianakos AP,et al. Altered expression of early cardiac markergenes in circulating cells of patients with hypertrophic cardiomyopathy. Cardiovasc Pathol 2007;16(6):329-335.
[124]Kontaraki JE,Marketou ME,Zacharis EA,et al.Early cardiac gene transcript levels in peripheral blood mononuclearcells in patients with untreated essentialhypertension.J Hypertens 2011;29(4):791-797.
[125]Kontaraki JE,Parthenakis FI,Patrianakos AP,et al. Myocardin gene regulatory variants as surrogate markers of cardiac hypertrophy-study in a genetically homogeneous population.Clin Genet 2007;73(1):71-78.
[126]Ransom JF,King IN,Garg V,et al.A rare human sequence variant reveals myocardin autoinhibition.J Biol Chem 2008;283(51):35845-35852.
[127]Kontaraki JE,Kochiadakis GE,Marketou ME,etal.Early cardiac gene transcript levels in peripheral blood mononuclear cells reflectseverity in stable coronary artery disease.Hellenic J Cardiol 2014;55(2):119-25.
[128]Torrado M,Centeno A,Lopez E,et al.In vivo forced expression of myocardin in ventricular myocardium transiently impairs systolic performance in early neonatal pig heart.Int J Dev Biol 2009;53(8-10):1457-1467.
[129]Torrado M,Iglesias R,Centeno A,et al.Targeted genesilencing reveals the functionalsignificance of myocardin signaling in the failing heart.PLo S One 2011;6(10): e26392.
[130]Tharp DL,Wamhoff BR,Turk JR,et al.Upregulation of intermediate-conductance Ca 2+-activated K+channel (IKCa1)mediates phenotypic modulation of coronary smooth mu scle.Am J Physiol Hea rt Circ Physiol 2006;291(5):H2493-H2503.
[131]Hendrix JA,Wamhoff BR,McDonald OG,etal.5ˊCArG degeneracy in smooth muscle alpha-actin is required for injury-induced gene suppression in vivo.J Clin Invest 2005;115(2):418-427.
[132]Talasila A,Yu H,Ackers-Johnson M,et al.Myocardin regulates vascular response to injury through miR-24/ -29a and platelet-derived growth factor receptor-beta. Arterioscler Thromb Vasc Biol 2013;33(10):2355-65.
[133]Tharp DL,Wamhoff BR,Wulff H,etal.Localdelivery of the KCa3.1 blocker,TRAM-34,prevents acute angioplasty-induced coronary smooth muscle phenotypic modulation and limits stenosis.Arterioscler Thromb Vasc Biol 2008;28(6):1084-1089.
[134]Shen J,Yang M,Jiang H,et al.Arterial injury promotes medial ch o n dro g en esis in Sm22 k n o ckout mice. Cardiovasc Res 2011;90(1):28-37.
[135]Minami T,Kuwahara K,Nakagawa Y,et al.Reciprocal expression of MRTF-A and myocardin is crucial for pathological vascular remodelling in mice.EMBO J 2012;31(23):4428-4440.
[136]Pfisterer L,Feldner A,Hecker M,et al.Hypertension impairs myocardin function:a novel mechanism facilitating arterial remodelling.Cardiovasc Res 2012;96(1): 120-9.
[137]Pfisterer L,Meyer R,Feldner A,et al.Bortezomib protects from varicose-like venous remodeling.FASEB J 2014;28(8):3518-27.
[138]Reynolds PR,Mucenski ML,Le Cras TD,et al.Midkine is regulated by hypoxia and causes pulmonary vascular remodeling.J Biol Chem 2004;279(35):37124-37132.
[139]Jie W,Wan g X,Hu an g L,et al.Co n trib ution of CXCR4(+)/PDGFRbeta(+)progenitor cells in hypoxic alveolar arterioles muscularization:role of myocardin. Cardiovasc Res 2010;87(4):740-750.
[140]Zhu P,Huang L,Ge X,etal.Transdifferentiation of pulmonary arteriolar endothelial cells into smooth musclelike cells regulated by myocardin involved in hypoxiainduced pulmonary vascular remod eling.Int J Exp Pathol 2007;87(6):463-474.
[141]Bell RD,Deane R,Chow N,et al.SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells.Nat Cell Biol 2009;11(2):143-153.
[142]Chow N,Bell RD,Deane R,et al.Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimerˊs phenotype.Proc Natl Acad Sci U S A 2007;104(3):823-828.
[143]Zlokovic BV.Neurovascularpathways to neurodegeneration in Alzheimerˊs disease and other disorders.Nat Rev Neurosci 2011;12(12):723-738.
[144]Zhou W,Negash S,Liu J,etal.Modulation of pulmonary vascular smooth muscle cellphenotype in hypoxia:role of cGMP-dependent protein kinase and myocardin.Am J Physiol Lung Cell Mol Physiol 2009;296(5):L780-L789.
[145]Jie W,Guo J,Shen Z,etal.Contribution of myocardin in the hypoxia-induced phenotypic switching of ratpulmonary arterial smo oth muscle cells.Exp Mol Path ol 2010;89(3):301-306.
[146]Latif N,Sarathchandra P,Chester AH,etal.Expression of smooth muscle cell markers and co-activators in calcified aortic valves.Eur Heart J 2014.
[147]Shats I,Milyavsky M,Cholostoy A,et al.Myocardin in tumor suppression and myofibroblast differentiation. Cell Cycle 2007;6(10):1141-1146.
[148]Chen F,Mo Y,Ding H,etal.Frequent epigenetic inactivation of Myocardin in human nasopharyngeal carcinoma.Head Neck 2011;33(1):54-59.
[149]Liao XH,Li YQ,Wang N,et al.Re-expression and epigenetic modification of maspin induced apoptosis in MCF-7 cells med iated by myocard in.Cell Sign al 2014;26(6):1335-46.
[150]Horiuchi A,Nikaido T,Taniguchi S,etal.Possible role of calponin h1 as a tumor suppressor in human uterine leiomyosarcoma.J Natl Cancer Inst 1999;91(9):790-796.
[151]Ramaswamy S,Ross KN,Lander ES,et al.A molecular signature of metastasis in primary solid tumors.Nat Genet 2003;33(1):49-54.
[152]PerotG,Derre J,Coindre JM,etal.Strong smooth muscle differentiation is dependent on myocardin gene amplification in most human retroperitoneal leiomyosarcomas. Cancer Res 2009;69(6):2269-2278.
[153]Malone MD,Kerr K,Kavanah M,etal.Primary leiomyosarcoma of the abdominal aorta.J Vasc Surg 1996;24(3): 487-493.
[154]Suga T,Iso T,Shimizu T,etal.Activation of receptor for advanced glycation end products induces osteogenic differentiation of vascular smooth muscle cells.J Atheroscler Thromb 2011;18(8):670-683.
[155]He SH,Wei AY,Yang Y,et al.Reduced expression of myocardin and serum response factor in the cavernous tissue ofdiabetic rats.Andrologia 2012;44(Suppl1):518-522.
[156]Wei AY,He SH,Zhao JF,et al.Characterization of corpus cavernosum smooth muscle cellphenotype in diabeticrats with erectile dysfunction.Int J Impot Res 2012;24(5): 196-201.
[157]He S,Zhang T,Liu Y,et al.Myocardin restores erectile function in diabetic rats:phenotypic modulation of corpus cavernosum smooth muscle cells.Andrologia 2014.
[158]Chen J,Chen H,Sanders KM,et al.Regulation of SRF/ CArG-dependentgene transcription during chronic partial obstruction of murine small intestine.Neurogastroenterol Motil 2008;20(7):829-842.
[159]Imamura M,Sugino Y,Long X,et al.Myocardin and microRNA-1 modulate bladder activity through connexin 43 expression during post-natal development.J Cell Physiol 2013;228(9):1819-26.
[160]Hermann J,Haas U,GressnerAM,etal.TGFb up-regulates serum response factor in activated hepatic stellate cells. Biochim Biophys Acta 2007;1772(11-12):1250-1257.
[161]Shimada H,Ochi T,Imasato A,et al.Gene expression profiling and functionalassays of activated hepatic stellate cells suggestthatmyocardin has a role in activation.Liver Int 2010;30(1):42-54.
[162]Morita T,Hayashi K.Arp5 is a key regulator of myocardin in smooth muscle cells.J Cell Biol2014;204(5):683-96.
[163]Joung H,Kwon JS,Kim JR,etal.Enhancer ofpolycomb1 lessens neointima formation by potentiation of myocardininduced smooth muscle differentiation.Atherosclerosis 2012;222(1):84-91.
[164]Hoggatt AM,Kim JR,Ustiyan V,et al.The transcription factor Foxf1 binds to serum response factor and myocardin to regulate gene transcription in visceralsmooth muscle cells.J Biol Chem 2013;288(40):28477-87.
[165]Liu ZP,Wang Z,Yanagisawa H,et al.Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and myocardin.Dev Cell 2005;9(2):261-270.
[166]Yin F,Herring BP.GATA-6 can act as a positive or negativ e regulator of smooth muscle-specific gene expression.J Biol Chem 2005;280(6):4745-4752.
[167]Zhou J,Hu G,Wang X.Repression of smooth muscle differentiation by a novel high mobility group box-containing protein,HMG2L1.J Biol Chem 2010;285(30): 23177-23185.
[168]Zhang SM,Gao L,Zhang XF,etal.Interferon regulatory factor 8 modulates phenotypic switching of smooth muscle cells by regulating the activity of myocardin.Mol Cell Biol 2014;34(3):400-14.
[169]Jiang DS,Luo YX,Zhang R,et al.Interferon regulatory factor 9 protects against cardiac hypertrophy by targeting myocardin.Hypertension 2014;63(1):119-27.
[170]Gordon JW,Pagiatakis C,Salma J,et al.Protein kinase A-regulated assembly of a MEF2-HDAC4 repressor complex controls c-Jun expression in vascular smooth muscle cells.J Biol Chem 2009;284(28):19027-19042.
[171]Lockman K,Taylor JM,Mack CP.The histone demethylase,jmjd1a,interacts with the myocardin factors to regulate SMC differentiation marker gene expression.Circ Res 2008;101(12):e115-e123.
[172]Nakamura S,Hayashi K,Iwasaki K,et al.Nuclear import mechanism for myocardin family members and their correlation with VSMC phenotype.J Biol Chem 2010; 285(48):37314-37323.
[173]Leenders JJ,Wijnen WJ,Hiller M,et al.Regulation of cardiac gene expression by KLF15,a repressor of myocardin activity.J Biol Chem 2010;285(35):27449-27456.
[174]Li HJ,Haque Z,Lu Q,etal.Steroid receptor coactivator 3 is a coactivator for myocardin,the regulator of smooth muscle transcription and differentiation.Proc Natl Acad Sci U S A 2007;104(10):4065-4070.
[175]Kiyan Y,Limbourg A,Kiyan R,etal.Urokinase receptor associates with myocardin to control vascular smooth muscle cells phenotype in vascular disease.Arterioscler Thromb Vasc Biol 2012;32(2):449-458.
[176]Tanaka T,Sato H,Doi H,etal.Runx2 represses myocardin-mediated differentiation and facilitates osteogenic conversion of vascular smooth muscle cells.Mol Cell Biol 2007;28(3):1147-1160.
[177]Zhou J,Zhang M,Fang H,etal.The SWI/SNF chromatin remodeling complex regulates myocardin-induced smooth muscle-specific gene expression.Arterioscler Thromb Vasc Biol 2009;29(6):921-928.
[178]Xu Z,Ji G,Shen J,etal.SOX9 and myocardin counteract each other in regulating vascular smooth muscle cell differentiation.Biochem Biophys Res Commun 2012;422(2): 285-90.
[179]Zhou J,Blue EK,Hu G,etal.Thymine DNA glycosylase represses myocardin-induced smooth muscle cell differentiation by competing with serum response factor for myocardin binding.J Biol Chem 2008;283(51):35383-35392.
[180]Madonna R,Willerson JT,Geng YJ.Myocardin Aenhances telomerase activities in adipose tissue mesenchymal cells and embryonic stem cells undergoing cardiovascularmyogenic differentiation.Stem Cells 2008;26(1):202-211.
[181]Martin E,Caubit X,Airik R,et al.TSHZ3 and SOX9 regulate the timing of smooth muscle cell differentiation in the ureter by reducing myocardin activity.PLoS One 2013;8(5):e63721.
[182]Xie C,Guo Y,Zhu T,et al.Yap1 protein regulates vascular smooth muscle cellphenotypic switch by interaction with myocardin.J Biol Chem 2012;287(18):14598-605.
✉Corresponding author:Joseph M.Miano,Ph.D,Aab Cardiovascular Research Institute,University of RochesterSchoolofMedicine and Dentistry, Rochester,New York,USA.E-mail:joseph_miano@urmc.rochester.edu.
Received 20 November 2014,Accepted 10 December 2014,Epub 25 December 2014
The author reported no conflict of interests.
©2015 by the Journal of Biomedical Research.All rights reserved.
10.7555/JBR.29.20140151
 THE JOURNAL OF BIOMEDICAL RESEARCH2015年1期
THE JOURNAL OF BIOMEDICAL RESEARCH2015年1期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Ventricular tachycardia ablation and substrate modification in ICD patients with electrical storm
- Induced pluripotentstem cells are induced pluripotentstem cell-like cells
- HLA antigens and anti-sperm antibody production in Iranian vasectomized men
- Unruptured pregnancy in a noncommunicating rudimentary horn at 37 weeks with a live fetus:a case report
- Surgicaloutcomes ofmini-open Wiltse approach and conventional open approach in patients with single-segment thoracolumbar fractures withoutneurologic injury
- Portalvein arterialization promotes liver regeneration after extended partialhepatectomy in a rat model