
杨树蛋白质磷酸化位点预测
2015-01-09 11:56高光芹黄家荣周俊朝谢鹏芳河南农业大学郑州450002
生物信息学 2015年3期
高光芹,黄家荣,周俊朝,谢鹏芳(河南农业大学,郑州450002)
doi:10.3969/j.issn.1672-5565.2015.03.04
杨树蛋白质磷酸化位点预测
高光芹,黄家荣∗,周俊朝,谢鹏芳
(河南农业大学,郑州450002)
以小黑杨磷酸化蛋白质组为研究对象,用人工神经网络表达丝氨酸、苏氨酸等残基位点的磷酸化与氨基酸序列的结构特征之间的非线性关系,建立了BP人工神经网络模型,并用磷酸化数据对所建模型进行训练和分析,得适宜的结构为21×16∶8∶4,拟合准确度为90%,Acc、Sn、Sp、MCC分别为78%、89%、67%、0.57,对比分析结果表明,所建模型具有较强的预测能力。
小黑杨;磷酸化蛋白质;磷酸化位点;人工神经网络
在生物体内,由RNA翻译形成的蛋白质都要经过翻译后修饰才具有生物活性,致使生物蛋白质分子具有某些功能位点、活性部位或功能结构域[1]。磷酸化位点是最重要的蛋白质功能位点,对细胞功能起着重要的调节作用。蛋白质磷酸化是在蛋白激酶催化作用下,磷酸基团由供体分子转移到蛋白质的含有羟基的氨基酸侧链上的过程,是一个可逆的过程,几乎调节着生命活动的所有过程。真核与原核生物的蛋白质磷酸化位点残基不同,前者主要是丝氨酸(Serine,S)、苏氨酸(Threonine,T)和酪氨酸(Tyrosine,Y)等残基;后者主要是天冬氨酸(Aspartic acid,D)、谷氨酸(Glycine,G)和组氨酸(Histidine,H)等残基。通过在磷酸化位点发生的酯化作用,改变蛋白质的结构、活性及其与其他分子相互作用的能力,在信号传导、基因表达、细胞分裂等许多生物学过程的调控中起着重要作用[2-3]。随着高通量鉴定磷酸化蛋白质技术的发展,尤其是质谱技术在蛋白质组学中的应用,磷酸化修饰数据不断积累,将计算方法引入磷酸化蛋白质组学的研究中,将有利于发现新的磷酸化修饰规律,并为生物学实验提供验证信息。现在,磷酸化位点预测方法,已从过去通过氨基酸序列预测发展出一系列新的算法[4-5],如人工神经网络,支持向量机等。当前,已有大量的蛋白质磷酸化位点预测研究[6-10],也有专门针对特定物种的蛋白质磷酸化位点预测分析[11],但针对杨树蛋白质磷酸化位点的预测研究尚未见报道。本文以小黑杨(Populus simonii×P nigra)磷酸化蛋白质组为研究对象,用人工神经网络构建磷酸化位点预测模型,为相关研究奠定基础。
1 材料与方法
1.1 样本收集与组织
从文献[12]鉴定提供的目前最大的木本植物磷酸化位点数据集下载小黑杨叶片蛋白质磷酸化位点(只有S和T残基)前后各10个氨基酸残基的序列106段,构成正样本集;再从拟南芥磷酸化数据库[13]按1∶1的比例下载对应的非磷酸化序列106段,构成负样本集,样式如表1。按样本集顺序每4个样本抽取1个(共54段)组成检验样本,剩下的3/4(共158段)作为训练样本。应用一种表2所示的新型氨基酸描述子[6,8]表征样本的氨基酸结构,并自编MATLAB程序,将字符串样本转换为数值样本。

表1 样本集样式Table 1 Type of sam ple set
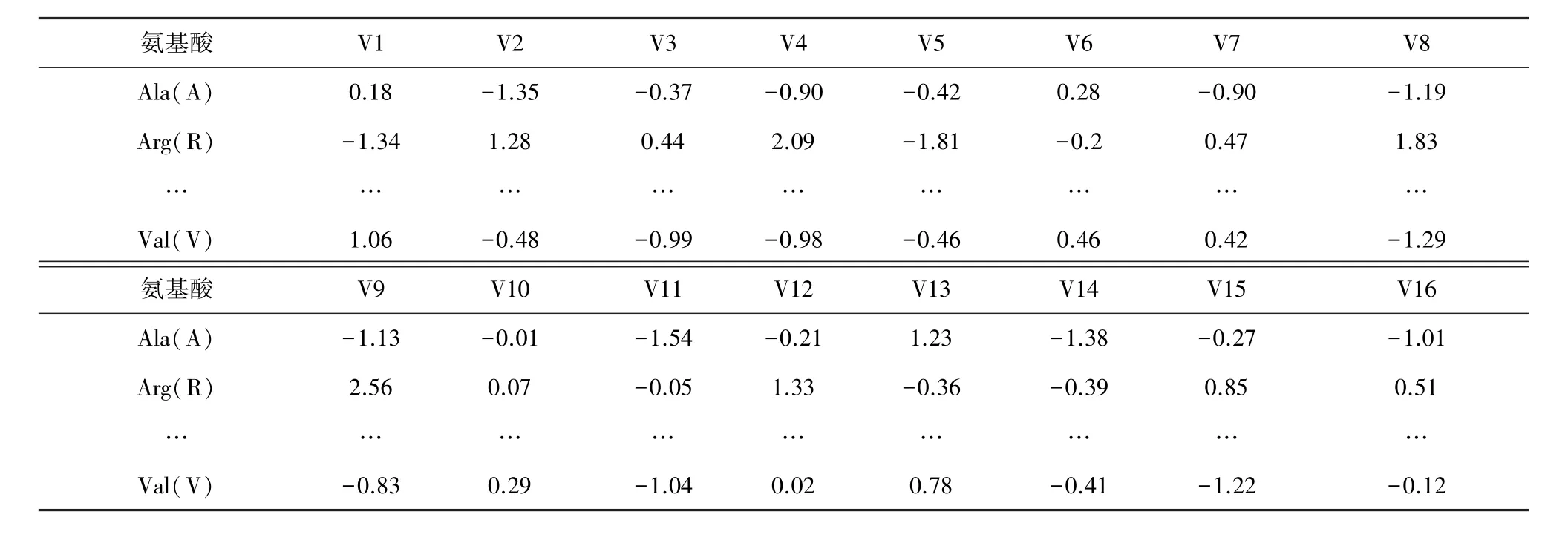
表2 氨基酸描述子V样式Table 2 Vscales for am ino acids
1.3 模型构建
以样本序列的21个氨基酸残基、每个残基m个描述子变量构成的21×m个V变量串联表征作为输入向量,以关系式n=log2m计算隐含层应取的神经元数,以样本序列的中心残基(S/T)是否被磷酸化构成的分类向量作为输出向量,构建了结构为21×m∶n∶4杨树蛋白质磷酸化位点神经网络预测模型。
模型的图形表达如图1,图中符号●、→、①、□、∫依次表示输入层节点、信息流、输入值为1的节点、神经元、对数S型作用函数。
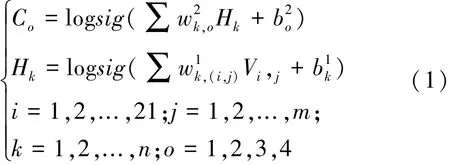
模型的数学表达为:式中,Co为输出层第o神经元的输出变量;Hk为隐层第k神经元的输出变量;Vi,j为输入层第i残基第j描述子节点的输k神入经变元量的;连接权表示输入层第(i,jk)节神点与隐层第;表示隐层第经元与输出层第o神经元的连接权;、分别为隐层第k神经元、输出层第o神经元的阈值;log sig()为MATLAB的对数S形函数。
1.4 模型训练与检验
在进行模型训练时,首先要在MATLAB系统中用氨基酸描述子对前面组织的字符串训练样本量化为数值样本。因数据量很大,用MATLAB语言编程进行处理。处理得到的训练样本——输入矩阵V和输出矩阵C分别是(21×m)×L和4×158的数值矩阵。将定义好的训练样本导入图形用户界面(GUI),并按图1进行网络设置后,就可进行神经网络模型的训练,最后将名为“network N”的网络对象等训练结果导出GUI并保存。模型的拟合性能检验,用测量学的精度计算方法;预测性能检验用生物信息学中常用的评价指标——准确率Acc、灵敏度Sn、特异度Sp、马修斯相关系数MCC[11,14],其算式如下:式中,TP—被正确分类的正(Positive)样本数目;TN—被正确分类的负(Negative)样本数目;FP—被错误分类的正样本数目;FN—被错误分类的负样本数目;T—总样本数目。L

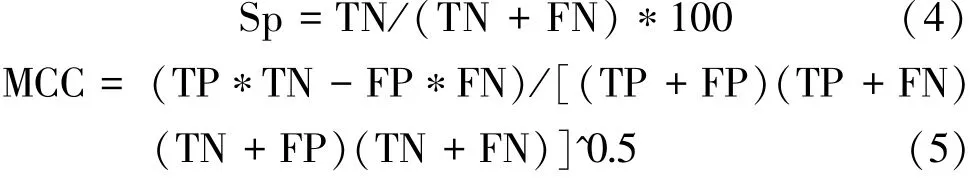
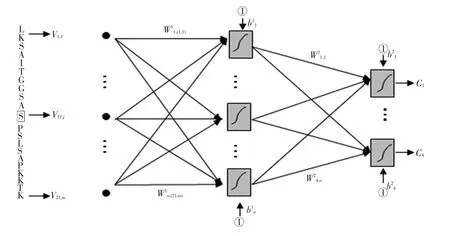
图1 杨树蛋白质磷酸化位点神经网络预测模型(21×m∶n∶4)Fig.1 Neural network model forecasting phosphorylation sites of pop lar protein(21×m∶n∶4)
2 结果与分析
以158段和54段氨基酸序列的描述子量化数据作为训练和检验样本,对所建模型按输入向量分为三种类型(Network1、Network2、Network3)进行训练、检验和对比分析(见表3),得最好的模型为network3,其结构为21×16∶8∶4,拟合准确度为90%,预测的正确率Acc、灵敏度Sn、特异度Sp、马修斯相关系数MCC等预测评价指标分别为78%、89%、67%、0.57。文献[5]用SVM研究的结果依次为74%,72%,77%,0.49;文献[6]基于SVM的氨基酸频率计算预测水稻蛋白质磷酸化位点的结果依次为75%,76%,67%,0.47。对比结果表明,除Sp指标外,其余指标都明显大于前人的研究结果,说明本文提出的模型network3也具有理想的蛋白质磷酸化位点预测能力。将network3重命名为NNFPSPP (Neural Network Forecasting Phosphorylation Site of Poplar Protein),其权值、阈值见表4,将其代入式(1),得杨树蛋白质磷酸化位点神经网络预测模型作用函数表达式,因输入变量和权值、阈值个数多,不便在此列出。在实际应用时,直接调用其MATLAB仿真函数表达式:

式中,sum()为MATLAB的仿真函数;NNFPSPP为训练好的网络对象,它储存了网络结构、属性等全部参数;V、C为模型的输入、输出向量。

表3 评价模型预测性能的指标Table 3 Indicators evaluating predict performance of themodel
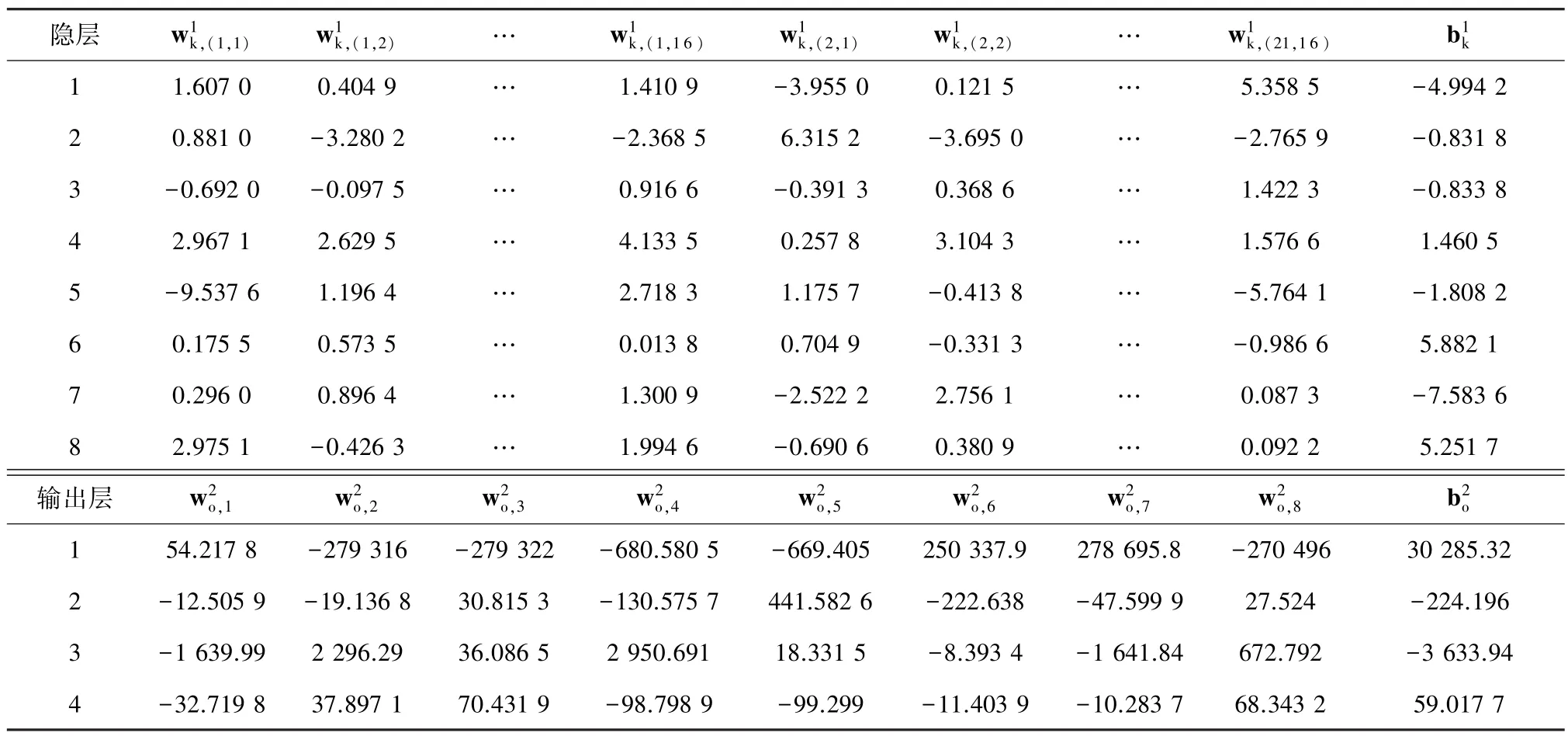
表4 NNFPSPP的训练结果Table 4 Straining results of NNFPSPP
3 结论与讨论
在论文的研究过程中,从磷酸化位点数据库下载、组建样本集是一个相当费事的工作,需要辅助于计算机的数据处理功能。我们自编的将字符串样本转换为数值样本的MATLAB程序,是一个有益的参考。
在准备好样本集的基础上,以样本序列的21个氨基酸残基、每个残基m个描述子变量构成的21xm个V变量串联表征作为输入向量,以关系式n=log2m计算隐含层应取的神经元数,以样本序列的中心残基(S/T)是否被磷酸化构成的分类向量作为输出向量,构建了结构为21×m∶n∶4杨树蛋白质磷酸化位点神经网络预测模型。
以158段和54段氨基酸序列的描述子量化数据为训练和检验样本,对所建模型按输入向量分三种类型(Network1、Network2、Network3)进行训练、检验和对比分析,得适宜的模型结构为21×16∶8∶4,模型的拟合准确度为90%,Acc、Sn、Sp、MCC分别为78%、89%、67%、0.57,除Sp指标外,其余指标值都明显优于前人的研究。
本文的研究特色:(1)针对木本植物建立蛋白质磷酸化位点预测模型,将林木生物信息资源作为森林资源的重要组成部分,将林木生物信息学纳入森林资源信息化管理研究,这对林学学科的发展将具有较大的促进作用;(2)将氨基酸序列片段与BP人工神经网络整合在一起的杨树蛋白质磷酸化位点神经网络预测模型,简单直观,通俗易懂,数形统一;(3)用MATLAB的仿真函数表达的预测函数式,是一种超常规数学表达,形式简捷,应用方便;(4)首次在木本植物中应用一种新型氨基酸描述子表征氨基酸性质与结构,使所建模型具有较强的预测能力。
应用的杨树蛋白质磷酸化实验数据,只有S、T的磷酸化,没有Y的磷酸化。这是否为杨树生物信息的特性之一,有待进一步验证。氨基酸性质与结构的新型描述子表征,可否明显提高杨树蛋白质结构预测的准确度[15],正作进一步研究。
前人对鉴定得到的磷酸化蛋白进行了细胞组件、分子功能及其所涉及的生物学途径分类研究[12],结果表明,小黑杨蛋白质的磷酸化广泛存在于细胞内的任何亚细胞结构,参与了几乎全部生命活动过程。由此可以按结构与功能的关系判定,所收集和组织的建模样本不会有很高的相似度,不会过高估计模型精度。
[1] 李伍举,吴加金.蛋白质功能位点预测[J].生物化学与生物物理进展,1993,20(1):60-62. LIWuju,WU Jiajin.Prediction of protein function site [J].Progress of Biochemistry and Biophysics,1993,20 (1):60-62.
[2] GLADIASM,TERESA F.Protein phosphorylation path⁃ways disruption by pesticides[J].Advances in Biological Chemistry,2013,3,460-474.
[3] ELLEN D,FREEK G B,DIDIER V,et al.Detection of cardiacmyosin binding protein⁃C(cMyBP⁃C)by a phos⁃pho⁃specific PKD antibody in contracting rat cardiomyo⁃cytes[J].Advances in Bioscience and Biotechnology,2013,4,1-6.
[4] QUE S,WANG Y,CHEN P,et a1.Evaluation of protein phosphorylation site predictors[J].Protein and Peptide Letters,2010,17:64-69.
[5] 胡敏菁,吴建盛,施识帆,等.面向蛋白质功能位点识别的机器学习平台构建[J].生物信息学,2010,8(1):12-15. HU Minjing,WU Jiansheng,SHI Shifan,et al.Machine learning platform for protein function sites prediction[J]. China Journal of Bioinformatics,2010,8(1):12-15.
[6] 李志良,李根容,舒茂,等.一种新型氨基酸拓扑结构信息矢量及在肽定量构效关系研究中的应用[J].中国科学B辑:化学,2008,38(8):745-754. LIZhiliang,LIGenrong,SHU Mao,et a1.A new type of amino acid topology information vector and application in research of peptide quantitative structure⁃activity relation⁃ship[J].China Science B:Chemistry,2008,38(8):745-754.
[7] 周鹏,周原,吴世容,等.一种基于三维原子场相互作用矢量的新型氨基酸结构信息描述子[J].科学通报,2008,51(1):34-39. ZHOU Peng,ZHOU Yuan,WU Shirong,et al.A new type of structure information descriptor for amino acid based on interaction vector in three dimensional atom field [J].Chinese Science Bulletin,2008,51(1):34-39.
[8] 舒茂.新型氨基酸结构表征方法及其在定量构效关系中应用研究[D].重庆:重庆大学,2009. SHU Mao.New Type of Characterization Method of Amino Acid Structure and its Application Research in Quantita⁃tive Structure⁃Activity Relationship[D].Chongqing:Chongqing University,2009.
[9] GAO J,THELEN J J,DUNKER A K,et al.Musite,a tool for global prediction of general and kinase specific phos⁃phorylation sites[J].Mol Cell Proteomics,2010,9(12):2586-600.
[10]NAKAGAMIH,SUGIYAMA N,MOCHIDA K,et al. Large⁃scale comparative hosphoproteomics identifies con⁃served phosphorylation sites in plants[J].Plant Physiolo⁃gy,2010,153:1161-1174.
[11]王伟,何华勤.基于SVM的氨基酸频率计算预测水稻蛋白质磷酸化位点[J].赤峰学院学报(自然科学版),2014,30(3):11-13. WANGWei,HE Huaqin.Prediction of rice protein phos⁃phorylation site based on amino acid frequency calculation with SVM[J].Journal of Chifeng University(Natural Sci⁃ence Edition),2014,30(3):11-13.
[12]刘晓羽.小黑杨叶片磷酸化蛋白质组及类囊体膜蛋白复合体的鉴定与分析[D].哈尔滨:东北林业大学,2010. LIU Xiaoyu.Identification and Analysis of Phosphopro⁃teome and Thylakoid Membrane Protein Complex in Leaf Blade of Populus[D].Harbin:Northeast Forestry Univer⁃sity,2010.
[13]HEAZLEWOOD J L,DUREK P,HUMMEL J,et al.Phos⁃PhAt:a database of phosphorylation sites in Arabidopsis thaliana and a plant⁃specific phosphorylation site predictor [J].Nucleic Acids Research,2007,36:D1015-1021.
[14]白海艳,吕军,张颖,等.蛋白质磷酸化位点的识别[J].内蒙古工业大学学报,2011,30(2):108-115. BAIHaiyan,LV Jun,ZHANG Ying,et al.Identification of protein phosphorylation sites[J].Journal of Inner Mon⁃golia University of Technology,2011,30(2):108-115.
[15]高光芹,孟庆玲,黄家荣.杨树蛋白质二级结构的人工神经网络预测[J].西北林学院学报,2014,29(5):59-63. GAO Guangqin,MENG Qingling,HUANG Jiarong.Predic⁃tion of poplar protein secondary structure with artificial neural networks[J].Journal of Northwast Forestry Univer⁃sity,2014,29(5):59-63.
Predicting phosphorylation sites of Poplar protein
GAO Guangqin,HUANG Jiarong∗,ZHOU Junchao,XIE Pengfang
(Henan Agricultural University,Zhengzhou 450002,China)
In this paper,the phosphoproteome of Populus simonii×P nigra was used as the research object.The nonlinear relationship between the structure characteristics of amino acid sequence and phosphorylation of serine and threoninewas expressed by artificial neural network.A BP artificial neural networkmodelwas established and trained by using the real data on phosphorylation.The appropriate structure is 21 x 16∶8∶4,the fitting accuracy is 90%,and the Acc,Sn,Sp,MCC are 78%,89%,67%,and 0.57,respectively.The comparative results show that the model has strong prediction ability.
Populus simonii×Pnigra;Phosphoproteome;Phosphorylation site;Artificial neural network
Q51
A
1672-5565(2015)03-165-05
2015-05-06;
2015-06-03.
河南省高等学校重点科研项目。
高光芹,女,硕士研究生,实验师,研究方向:化学生物信息学;E⁃mail:sckdggq@163.com.
∗
黄家荣,男,博士,教授,研究方向:森林资源信息化管理;E⁃mail:huangjiarong137@163.com.
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
天津医科大学学报(2019年6期)2019-08-13
时代英语·高一(2019年1期)2019-03-13
分析化学(2017年12期)2017-12-25
喜剧世界(2017年10期)2017-06-01
池州学院学报(2015年3期)2016-01-05
安徽医科大学学报(2015年9期)2015-12-16
天津科技大学学报(2015年2期)2015-08-09
遗传(2014年3期)2014-02-28
