
QuEChERS 法结合高效液相色谱-串联质谱法测定保健食品中12 种双酚类化合物
2014-12-26 01:58高梦婕盛永刚赵善贞邓晓军郭德华丁卓平王国民
色谱 2014年11期
高梦婕, 周 瑶, 盛永刚, 赵善贞, 邓晓军* ,郭德华, 丁卓平* , 王国民, 彭 涛
(1. 上海海洋大学食品学院,上海201306;2. 上海出入境检验检疫局,上海200135;3. 重庆出入境检验检疫局,重庆400020;4. 中国检验检疫科学研究院,北京100123)
双酚类化合物属内分泌干扰物,对人类和生物有毒害作用[1,2]。双酚类化合物存在于塑料包装材料中,在食品的加工运输贮藏过程中会迁移至食品内容物中,进而危害人体健康[3-6]。随着保健食品业快速发展,保健食品的日常食用量越来越大,食品接触包装材料中双酚类化合物的迁移对人体的影响越来越受到重视。双酚A(BPA)及环氧化物、双酚F(BPF)及环氧化物对生殖系统发育有很大危害,并严重影响胎儿及儿童的大脑发育[4,5];双酚S(BPS)可作为双酚A 的代用品,同时是医用高分子材料及各种塑料的重要原料,具有扰乱生殖系统的危害[6]。
目前,对于双酚类化合物的迁移限量规定并不完善。欧盟法规(EC/1895/2005 号指令)规定自2006 年1 月1 日以后禁止在食品接触的涂料中使用双酚F 二缩水甘油醚(BFDGE),并规定双酚A二缩水甘油醚(BADGE)、双酚A-(2,3-二羟基丙基甘油醚)(BADGE-H2O)、双酚A-双(2,3-二羟基丙基醚)(BADGE-2H2O)在食品或食品模拟物中的迁移总量不超过9 mg/kg,单一各类在食品或食品模拟物中的迁移总量不超过1 mg/kg[7]。同时欧盟塑料法规[8]和我国国家标准[9]均规定双酚S 在食品或食品模拟物中的迁移限量为0.05 mg/kg。
双酚类化合物的检测方法主要有高效液相色谱法和液相色谱-质谱联用法[10-13]。前者对于复杂基质中痕量残留物质的分析不能提供结构方面的信息,故在实际残留分析中受到一定限制。对于样品前处理而言,传统的固相萃取技术溶剂使用量大、步骤繁琐且耗时耗力。而QuEChERS 法具有快速、简单、便宜、有效、可靠和安全等特点[14,15],适合基质复杂的保健食品。目前,对于双酚类化合物的检测仅限于罐头产品的研究[10-12],还未见保健食品中同时检测12 种双酚类化合物的报道。本文通过改进的QuEChERS 法进行前处理,高效液相色谱-串联质谱(HPLC-MS/MS)同时测定保健食品中12 种双酚类化合物,操作简单、耗时短、应用前景广泛。
1 实验部分
1.1 仪器与试剂
API 4000 型四极杆串联线性质谱仪(美国AB公司);UFLC XR 高效液相色谱仪(日本岛津公司);电子分析天平(精度为0.000 1 g,德国Sartorius ME 公司);XW-80A 涡旋混合器(上海医科大学仪器厂);Milli-Q 超纯水一体机(Millipore 公司);NEVAP 氮吹仪(美国Organomation 公司);Allegia X-22R 高速冷冻离心机(Beckman 公司);0. 22 μm有机相滤膜(上海安谱科学仪器有限公司)。
BPA 和BPF 标准品(纯度>95%,日本TCI 公司);BFDGE 标准品(纯度>95%,德国Dr. Ehrenstorfer 公司);双酚S 标准品(纯度>99%,美国Admas公司);其余8 种双酚类化合物标准品(纯度>99%,瑞士Fluka 公司)。乙腈、甲醇、正己烷(ACS 纯,美国Merck 公司);甲酸、乙酸(ACS 纯,J.T. Baker 公司);乙酸铵(分析纯,上海国药集团);NaCl、无水MgSO4、无水Na2SO4和乙酸钠(中国医药上海化学试剂公司);CNW-BONDN-丙基乙二胺(PSA)、石墨化炭黑(GCB)和高碳C18(HC-C18)粉末(上海安谱公司);实验用水为超纯水(Millipore系统生产)。
标准溶液的配制:分别准确称取0.010 0 g 标准品于10 mL 容量瓶中,用甲醇溶解并定容至刻度,4℃下保存。12 种双酚类化合物标准储备液质量浓度为1.000 g/L,用甲醇稀释为10 mg/L 混合标准溶液,于4 ℃避光保存。取空白样品基质按照1.2 节步骤制备得基质空白溶液,加入适量混合标准溶液,配制成适当浓度的基质匹配标准溶液,临用时现配。
1.2 样品前处理
1.2.1 固体粉状和口服液类保健食品
称取均质样品2 g,置于50 mL 离心管中,加入适量双酚类标准溶液、4 mL 去离子水、7.5 mL 含0.1%(v/v)乙酸乙腈溶液、1.5 g 醋酸钠和1 g NaCl,超声混合5 min,以5 000 r/min 离心5 min,收集上清液;基质中加入7.5 mL 含0.1% (v/v)乙酸乙腈溶液进行二次提取,合并两次上清液。加入100 mg PSA、50 mg GCB、50 mg HC-C18和1 g 无水Na2SO4,涡漩混合5 min,以5 000 r/min 离心5 min;移取上清液5 mL 至玻璃管中,在40 ℃下氮气吹干,用1 mL 含0.1% (v/v)甲酸的甲醇-水(50∶50,v/v)复溶;溶液转移至1.5 mL 离心管中,以10 000 r/min 离心5 min;上清液过0.22 μm 滤膜,待测。
1.2.2 胶囊类保健食品
称取胶囊中的内容物1 g,置于50 mL 塑料离心管中,加入适量双酚类标准溶液、4 mL 乙腈饱和正己烷,涡漩混合3 min,加入5 mL 正己烷饱和乙腈,超声混合5 min,以5 000 r/min 离心5 min,收集下层清液;基质中加入5 mL 正己烷饱和乙腈进行二次提取,合并两次的下层清液。加入100 mg PSA、200 mg HC-C18、1 g 无水Na2SO4,涡漩混合5 min,以4 000 r/min 离心10 min;移取下层清液5 mL 至玻璃管中,在40 ℃下氮气吹干,用1 mL 含0.1%(v/v)甲酸的甲醇-水(50∶50,v/v)复溶;溶液转移至1.5 mL 离心管中,以10 000 r/min 离心5 min;上清液过0.22 μm 滤膜,待测。
1.2.3 实际样品的测定
实际样品不加入双酚类标准物质,不同基质的样品前处理分别按照1.2.1 节和1.2.2 节进行。
1.3 色谱-质谱条件
Thermo Aquasil C18色谱柱(150 mm×4.6 mm,3.0 μm)。流动相A 和B 分别为甲醇和0.1% (v/v)甲酸溶液(含0.005 mol/L 醋酸铵);进样量:10 μL;流速:0.6 mL/min;柱温:30 ℃。梯度洗脱程序为:0 ~5 min,50% A;5 ~12 min,50% A ~80% A;12 ~19 min,80% A;19 ~21 min,80% A ~50% A;21 ~22 min,50%A。
离子源:电喷雾离子化正离子模式(ESI+)和负离子模式(ESI-);扫描方式:多反应监测(MRM);电喷雾电压(IS):5 500 V;雾化气压力(GS1):413.8 kPa;气帘气压力(CUR):172.4 kPa;辅助气压力(GS2):413.8 kPa;离子源温度(TEM):500℃;碰撞活化解离(CAD):高(high)。12 种双酚类化合物的质谱参数见表1。

表1 测定12 种双酚类化合物的质谱参数Table 1 Parameters of MS/MS for the determination of the 12 bisphenol compounds
2 结果与讨论
2.1 流动相的选择
比较了甲醇-水和乙腈-水流动相中加入一定量的醋酸铵和甲酸等添加剂对目标化合物离子化效率的影响。在流动相中加入0.005 mol/L 醋酸铵有助于消除钠盐的干扰,而加入0.1%(v/v)甲酸有助于待测物母离子峰的形成[16]。实验结果显示,以甲醇为有机相时,各待测物响应强度较稳定,且峰形均较好;而乙腈为有机相时,响应值明显降低,分离度也不佳,不适合同时检测。故选用对双酚类具有较好分离效果的甲醇为有机相。初始流动相比例为甲醇-水(50∶50,v/v),在10 min 内甲醇的体积分数达到80%,可完全洗脱所有双酚类物质,出峰明显,故洗脱时间设定为22 min。12 种双酚类化合物的多反应监测色谱图见图1。
2.2 质谱条件的优化
取1.0 mg/L 的双酚类化合物标准溶液,分别以流动注射的方式在ESI+和ESI-模式下进行电离监测,结果发现:BPA 和BPF 在ESI-模式下有更好的响应,其余10 种双酚类化合物在ESI+模式下有更好的响应。再将各物质在ESI 模式下进行母离子扫描,BPA 和BPF 在一级全扫描质谱图中得到[MH]-,其余10 种双酚类化合物在一级全扫描质谱图中得到[M +NH4]+;参照国际食品法典委员会(CAC)和EU 657/2002/EEC 号决议中有关规定[17],选择两对离子进行MRM 监测即可,应选择实际样品分析中基质干扰较少的离子对,并考虑每种化合物标准品的质谱图和结构特性。通过调节仪器参数使母离子的丰度最大,进一步对子离子进行二级质谱扫描,得到碎片离子信息,选取丰度较强、干扰较小的两对子离子作为定性、定量离子,并优化去簇电压(DP)、碰撞电压(CE)等参数条件,以达到最佳灵敏度。12 种双酚类化合物的质谱参数见表1。

图1 12 种双酚类化合物的多反应监测色谱图Fig.1 MRM Chromatograms of the 12 bisphenol compounds
2.3 前处理条件的优化
2.3.1 提取液的优化
双酚类化合物的结构中均含两个及以上的苯环,易溶于乙腈、丙酮、甲醇等有机溶剂。同时,乙腈中加入酸有利于提高双酚类化合物的提取效率[11]。比较了含不同体积分数(0.1%、1%)的甲酸或乙酸的乙腈溶液对待测物的提取效果。图2 结果显示,采用含1%乙酸的乙腈溶液可从基质中充分提取目标化合物,故最终选定含1% 乙酸乙腈溶液作为提取溶剂。

图2 使用不同溶剂的前处理方法对待测物的回收率Fig.2 Recovery of analytes by various solvents in the process of preparation
2.3.2 净化剂的优化
在处理保健食品的基质净化时,以配方奶粉、口服液和胶囊类为基质。QuEChERS 方法的净化剂种类有很多,不同的净化剂发挥不同的作用。PSA主要用于吸附样品中的糖类和有机酸等弱酸性成分,GCB 用于去除样品中的色素,而HC-C18对去除样品中的蛋白质和脂类成分有较好的效果[18,19]。根据保健食品不同基质的特点,比较了PSA、GCB和HC-C18的组合对净化和回收的影响(见表2)。基质为固体粉状和口服液时,净化剂最优组合为PSA 100 mg、GCB 50 mg、HC-C1850 mg。胶囊类样品的基质中几乎不含色素,不加入GCB;其干扰主要来自脂肪,需加入HC-C18去除,故净化剂最优组合为PSA 100 mg、HC-C18200 mg。
2.4 方法学验证
2.4.1 线性范围与检出限
选取空白基质溶液配制12 种双酚类化合物的混合标准溶液,以峰面积和对应的含量作图。结果表明,12 种化合物在0.5 ~50.0 μg/kg 范围内线性关系良好,相关系数(r)均大于0.99(见表3)。以S/N >10 和S/N >3 确定12 种双酚类化合物的定量限(LOQ)及检出限(LOD)分别为0.4 ~1.7 μg/kg 及0.1 ~0.5 μg/kg,优 于 文 献[11]结 果(BADGE-HCl、BADGE-2HCl 和BFDGE-2H2O 的LOQ 为2.0 μg/kg)。

表2 净化步骤中净化剂组合的优化结果(n =6)Table 2 Results of optimal combination of clean-up procedure (n =6)

表3 12 种双酚类化合物的相关系数(r)、检出限及定量限(n =6)Table 3 Correlation coefficients (r),limits of detection (LODs)and limits of quantitation (LOQs)of the 12 bisphenol compounds (n =6)
2.4.2 回收率与精密度
选取不含双酚类化合物的配方奶粉、口服液和鱼油胶囊为空白基质,添加12 种双酚类化合物混合标准溶液,进行低、中、高3 个水平的加标回收试验。每个添加水平各取6 个独立样品进行处理,考察方法的回收率和相对标准偏差(见表4)。本方法对12 种双酚类化合物有良好的准确度与精密度。
最低添加水平(2.0 μg/kg)和空白口服液样品的MRM 色谱图见图3。
2.4.3 基质效应
液相色谱-质谱进行检测时,由于基质效应的影响,目标化合物发生离子增强或抑制作用[20]。
采用不含待测化合物的配方奶粉、口服液和鱼油胶囊的基质空白溶液添加100 μg/kg 的双酚类化合物,平行进样3 次,按照公式(1)对其峰面积进行归一化:
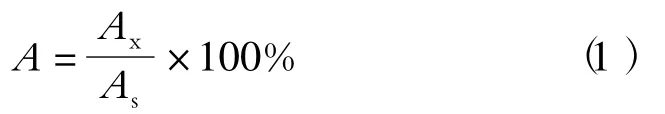
其中Ax为某基质中添加的双酚类化合物的峰面积,As为双酚类化合物标准溶液的峰面积。结果表明,基质抑制率在47.1% ~116.4% 之间,特别是在鱼油胶囊和配方奶粉中存在较强的基质效应,可能是由于化合物竞争离子化造成的[21]。本实验采用基质曲线进行定量分析,可以减弱基质效应的影响。
2.5 实际样品的测定
采用本方法对市售的100 种不同类型的保健食品进行测定,12 种双酚类化合物在胶囊中均未检出;在配方奶粉、固体饮料和口服液中检出BPA、BADGE-2H2O、BADGE-2HCl、BFDGE,含量为0.8~29.6 μg/kg。说明双酚类化合物在有些保健食品的包装材料中存在,并迁移到食品内容物中,但含量相对较低,未超过欧盟对食品中迁移总量的规定。

图3 (a)加标(2.0 μg/kg)和(b)空白口服液样品的MRM 色谱图Fig.3 MRM chromatograms of (a)spiked (2.0 μg/kg)and (b)blank oral solution samples

表4 12 种双酚类化合物的添加回收率及相对标准偏差(n =6)Table 4 Spiked recoveries and RSDs of the 12 bisphenol compounds (n =6)
3 结论
建立了采用QuEChERS 法结合高效液相色谱-串联质谱分析保健食品中12 种双酚类化合物的方法,并应用于实际样品的检测。该方法操作简便、快速、灵敏度高、重现性好、回收率佳,满足现行法规要求,可实现保健食品中双酚类化合物的定性定量,具有实际应用价值。
[1] Cabado A G,Aldea S,Porro C,et al. Food Chem Toxicol,2008,46:1674
[2] Miao J Z,Xue M,Zhang H. Chinese Journal of Analytical Chemistry (缪佳铮,薛鸣,张虹. 分析化学),2009,37(6):911
[3] Cao G Z,Chen S H,Xiao D Q,et al. Chinese Journal of Analytical Chemistry (曹国洲,陈少鸿,肖道清,等. 分析化学),2014,42(3):403
[4] Bao Y,Wang H Y,Li Z Q,et al. Food Science (鲍洋,汪何雅,李竹青,等. 食品科学),2011,32(21):261
[5] Hu X W,Zhang W D,Liu Y Q. Food Science (胡向蔚,张文德,刘炎桥. 食品科学),2006,27(4):264
[6] Cao G P,Wang B B,Ding Q C,et al. Chemistry &Bioengineering (曹桂萍,王蓓蓓,丁其晨,等. 化学与生物工程),2010,27(10):86
[7] Commission Regulation EC/1895/2005
[8] Commission Regulation EU No.10/2011
[9] GB 9685-2008
[10] Zhao X Y,Fu X F,Wang P,et al. Chinese Journal of Chromatography (赵晓亚,付小芳,王鹏,等. 色谱),2012,30(10):1002
[11] Liang K,Deng X J,Yi X H,et al. Chinese Journal of Analytical Chemistry (梁凯,邓晓军,伊雄海,等. 分析化学),2012,40(5):705
[12] Zhang Z H,Luo S L,Wu S L,et al. Journal of Instrumental Analysis (张朝晖,罗生亮,吴少林,等. 分析测试学报),2009,28(6):714
[13] Xiao D Q,Liu Z M,Ma M,et al. Journal of Instrumental Analysis (肖道清,刘在美,马明,等. 分析测试学报),2013,32(12):1502
[14] Lehotay S J,Mastovska K,Yun S J. J AOAC Int,2005,88(2):630
[15] Xu R,Wu J W,Liu Y G,et al. Chemosphere,2011,84:908
[16] Guo D H,Deng X J,Zhao S Z,et al. Chinese Journal of Analytical Chemistry (郭德华,邓晓军,赵善贞,等. 分析化学),2010,38(3):318
[17] European Union (2002/657/EC)
[18] Angelika W,Marek B. Food Chem,2011,125:803
[19] George S,Timothy B. Anal Chim Acta,2009,637:68
[20] Xiang P,Shen M,Zhuo X Y. Journal of Instrumental Analysis(向平,沈敏,卓先义. 分析测试学报),2009,28(6):753
[21] Little J L,Wempe M F,Buchanan C M. J Chromatogr B,2006,833(2):219
猜你喜欢
塑料包装(2021年3期)2021-07-20
中成药(2018年12期)2018-12-29
基层中医药(2018年7期)2018-12-06
知识经济·中国直销(2018年11期)2018-11-26
中成药(2018年1期)2018-02-02
中成药(2017年8期)2017-11-22
中成药(2017年3期)2017-05-17
中国塑料(2016年4期)2016-06-27
中国塑料(2016年2期)2016-06-15
中国卫生(2014年2期)2014-11-12
