
制药企业GMP文件生命周期管理调查Δ
2014-12-03 03:06浙江医药高等专科学校浙江宁波315100
中国药房 2014年5期
丁 静(浙江医药高等专科学校,浙江宁波 315100)
据国家食品药品监督管理总局(CFDA)统计,截至2013年7月,我国通过2011年版《药品生产质量管理规范》(GMP)认证的无菌药品生产企业仅占25.9%,非无菌药品生产企业仅占13.4%[1]。可见,我国制药企业GMP整体认证通过率较低,认证压力较大。国家食品药品监督管理部门曾明确规定,全国所有无菌药品生产企业应在2013年12月31日前达到2010年版GMP认证要求,而非无菌药品生产企业应在2015年12月31日前达到2010年版GMP认证要求。届时,那些未通过2010年版GMP认证的企业,将一律被要求停产,而先行通过2010年版GMP认证的企业可获得多方面扶持。因此,所有制药企业都应当抓住有利时机,积极应对挑战,除开展硬件设施方面的改造外,还应建立一套系统、科学、实用的文件管理系统,并持续、有效地开展GMP文件生命周期管理,以顺利通过2010年版GMP认证。本文拟就如何进行GMP文件生命周期管理以符合2010年版GMP要求等问题进行探讨和分析。
1 GMP文件生命周期管理模型简介
“文件生命周期”这一概念最早由美国档案学者菲利普·布鲁克斯提出。文件生命周期理论是指文件从产生直至因丧失作用被销毁或因具有长远历史价值被档案馆永久保存的整体运动过程[2]。该理论为文件的全过程管理奠定了理论基础,GMP文件管理在一定程度上体现了该理论的思想。但是,由于该理论主要是针对档案馆的文件管理,因此并不完全适用于制药企业的GMP文件管理。为此,笔者在该理论的基础上,结合GMP文件管理的特殊性,提出了GMP文件生命周期管理模型(见图1)。该模型覆盖了GMP文件整个生命运动的过程和形态,体现了文件的生命是“往复运动于起草到处置的连续的过程”,反映了GMP文件从起草、审核、批准、分发、培训、生效、执行、保管、复审、变更、替换、撤销、销毁等完整的运动过程,强调GMP文件的整体性、系统性、连贯性和运动性。该模型对指导和解释我国制药企业文件管理中的一些实际问题具有重要的现实意义。开展GMP文件生命周期管理对提升制药企业的质量管理水平、促进GMP的有效实施和推动我国制药工业持续健康发展具有举足轻重的作用。
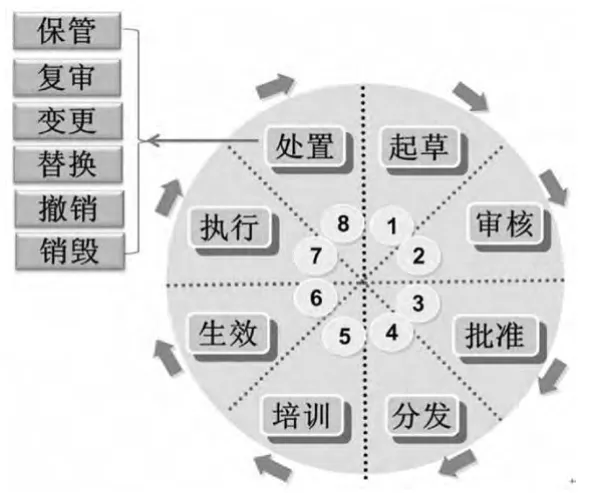
图1 GMP文件生命周期管理模型Fig 1 GMPdocument lifecycle management model
2 对象与方法
2.1 调查目的
调查我国制药企业的GMP文件管理情况,分析在GMP文件生命周期管理中存在的问题,从而有针对性地提出对策,提升其质量管理水平。
2.2 调查对象
选取浙江地区宁波4家、金华2家和嘉兴、绍兴、台州各1家制药企业作为调查对象,通过实地调研、专家访谈、企业资料查阅等相结合的方式展开调查。其中,访谈对象主要包括企业的质量管理负责人、质量管理员、办公室人员、一线操作工和药品监管部门人员。调查时间为2012年5月-2013年5月。
2.3 调查方法
按照制药企业在GMP文件起草、审核、批准、分发、培训、生效、执行、保管、复审、变更、替换、撤销及销毁等方面的情况进行分类调查,分析存在的问题,并采用Excel 2010软件进行数据整理和分析。
3 制药企业GMP文件生命周期管理中存在的问题
文件是确保信息有效传递和管理的工具[3]。GMP文件应该是贯穿于药品生产管理全过程的系统全面、规范完整、相互协调、操作性强的有机体系。若缺少文件,企业的质量风险将会大大增加;相反,完善的文件系统能够最大程度地降低风险[4]。笔者通过对以上9家制药企业进行调研,发现其在GMP文件生命周期管理中存在一些普遍的问题。GMP文件生命周期各阶段存在问题的企业数量统计见图2;GMP文件生命周期管理问题鱼骨图见图3。
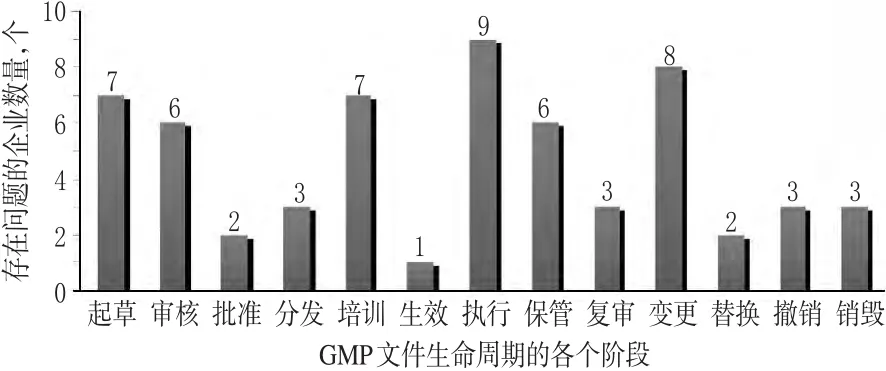
图2 GMP文件生命周期各阶段存在问题的企业数量统计Fig 2 The number of enterprises existing problems at various stages of GMPdocument lifecycle
3.1 文件起草缺乏系统性、完整性、可操作性及扩展性
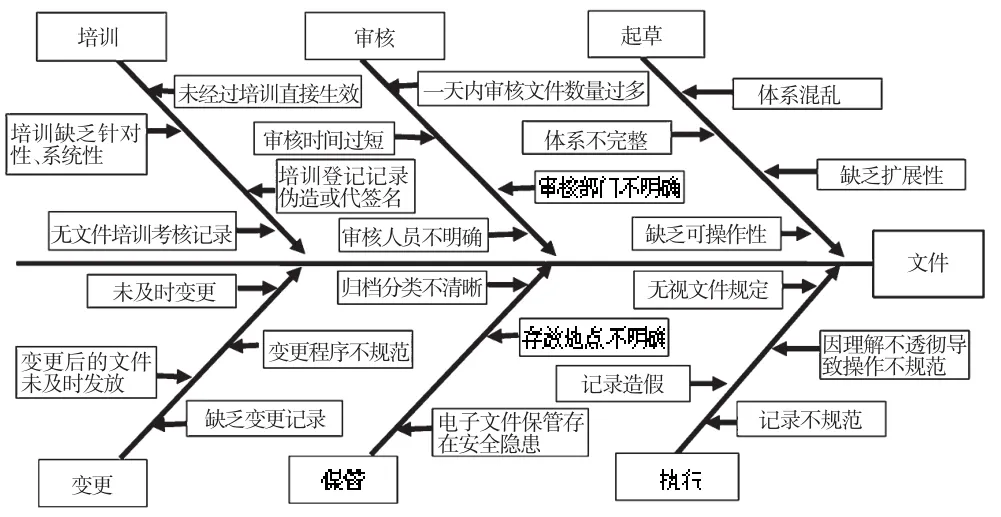
图3 GMP文件生命周期管理问题鱼骨图Fig 3 Fishbone diagram of GMP document lifecycle management issues
文件起草是GMP文件生命周期管理的重要开端,直接关系到后续GMP文件审核、批准、执行等一系列活动。经笔者调研发现,不少制药企业在起草文件时缺乏整体设计,具体表现在以下方面:(1)文件不成体系或体系混乱。部分企业的文件格式不统一,内容缺乏系统性、协调性;部分企业存在真假两套文件,一套仅用于应付检查,并无实际的指导意义。(2)文件体系不完整。部分企业的图纸、标签、货位卡等无文件编号,为不可控文件;部分企业尚未制订如风险管理、变更管理等2011年版GMP新增的文件。(3)所制订文件缺乏可操作性。文件起草人员在起草文件时过多地依赖网络资源,而对该企业的实际情况调查不足,导致起草的文件与企业实际生产环境、运行模式脱节。文件制订不够具体和细化,部分文件的操作参数不明确,有的参数范围偏大,且不少参数的设定未经过充分验证。(4)文件缺乏扩展性,不适应企业发展要求。
3.2 文件审核形式化
文件审核是GMP文件生命周期管理中的一个重要环节。通过审核,能够发现文件在起草中存在的问题,确保文件的规范性、科学性和兼容性。在调研过程中,笔者查看了多家企业的GMP文件的审核记录,发现存在以下问题:(1)同一审核人在一天内审核文件数量过多,审核时间过短,难以发现文件在起草中存在的诸如缺乏系统性、兼容性、可操作性等问题,文件审核成为应付监管部门检查的一项形式性工作。(2)文件审核部门、审核人员不明确。部分企业的GMP文件未经质量管理部门审核,而部分企业的GMP文件审核人员多达十余个,审核基本只是签名。
3.3 对相关人员的培训效果不佳
依据GMP文件生命周期模型,文件批准后,将由文件起草人或审批人对相关部门的相关人员进行培训后,文件才能够正式生效。培训的目的是使每个岗位的员工都能正确理解和掌握与本岗位有关的文件,能按照文件规定规范地、标准地开展工作。在调研中,笔者发现在文件生效前,某些企业存在未对相关人员进行培训,培训缺乏针对性、系统性,培训登记记录伪造或代签名、无培训考核记录等问题。员工对文件内容或是一无所知,或是一知半解,直接影响到了文件的执行。
3.4 文件执行不到位
文件执行是GMP文件生命周期管理中最为重要的环节,是GMP管理的落脚点,直接关系到企业质量管理体系的有效性、真实性。从调查情况看,几乎所有的企业在文件执行上都存在或多或少的问题,主要包括:(1)无视文件规定。如物料未经质量评价即批准放行。(2)对标准操作规程(SOP)理解不透彻,导致操作不规范。如某企业员工对洁净区器具清洗SOP不理解,未严格按照SOP进行操作,造成器具清洗不彻底,引起交叉污染。(3)部分企业的偏差管理等文件虽然制订完美,但并未能真正有效执行。如某生产批次的成品率仅为59%,而平均成品率是90%,被错误列为微小偏差,并未进行充分调查和详细记录。(4)记录造假。如记录中显示某日已开启使用某仪器,但实际当天并未开启。(5)记录不及时、不清晰、不完整,任意涂改和撕毁记录,不按规定签名和审核,直接影响到了产品质量的追溯和事故原因的调查。
3.5 文件处置不当
依据GMP文件生命周期管理模型,GMP文件生命周期管理的第八阶段是文件处置,具体包括文件保管、复审、变更、替换、撤销、销毁等情况。调查发现,一些企业存在的主要问题包括:(1)文件归档分类不清晰,存放地点不明确,查找效率低下。(2)电子文件保管存在安全隐患。不少企业的电子文件可随意更改、复制、删除,且并未进行备份,难以保证其原始性、真实性、准确性和安全性。(3)文件变更控制不完善。一方面,文件未及时变更,如某企业的一台设备已报废3年,但相应的设备操作规程等文件并未进行变更;另一方面,文件变更程序不规范。部分企业变更文件只在原文件上划改,或未经审批直接替换原文件;变更后的文件未及时发放到使用者手中,导致变更后的文件未能有效执行。
4 完善GMP文件生命周期管理的对策
4.1 顶层设计文件
4.1.1 工作流程科学化,文件体系统一化。企业可依据GMP文件生命周期管理思想,建立科学、合理的文件管理操作规程,实现工作流程科学化。质量管理部门明确文件分类和编号,确定本企业文件的统一格式,可避免随意性和盲目性,实现文件体系统一化。调研发现,某些企业的文件表头过于简单,并不能充分反映该文件从起草到销毁的完整的生命周期运动过程。为此,笔者设计了GMP文件表头供企业参考,详见图4。
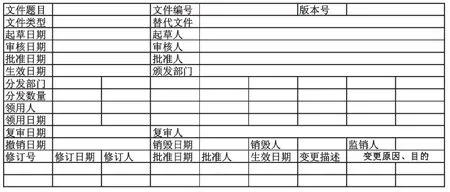
图4 GMP文件表头Fig 4 File header of GMP
4.1.2 文件起草团队化。GMP文件起草团队成员须经过GMP培训并考核合格,全面掌握GMP的各项要求,熟悉本专业的技术和管理,实践经验丰富,并有较强的文件撰写能力。文件起草需要在企业质量管理负责人的指导下进行,实行公司高层、工艺技术人员、管理人员和一线操作人员四方结合,明确责任分工,相互沟通协调。
4.1.3 文件内容专业化。①理解GMP要求,整体设计GMP文件。要做到文件目录无遗漏、无重叠,文件系统内部互相协调一致,不得出现同一操作在不同的文件中相互矛盾的情况[5]。②符合企业实际,增强可操作性。文字表述准确、规范,不得模棱两可,可量化的必须量化,相关参数需经过验证,做到科学、严谨。③图文并茂,清晰易懂。尤其是SOP,要尽量使用表格、清单和流程图,以便于一线操作人员掌握。
4.2 明确职责,加强文件审批
文件审核人员需经过GMP培训,具有较全面的专业知识,实践经验丰富,善于发现、纠正文件中的错误及不完善的地方,具有一定的决策、平衡和协调能力,具有严谨、认真、负责任的工作态度。建议企业预先设定具体文件的审核人,制订文件审核职责对照表,明确审核部门和审核人,避免权责不清、相互推诿。文件审批职责应写入部门职责、岗位职责中,并落实到人。此外,文件审核人员应控制单次审批的文件数量,留足充分的时间用于仔细审核文件,确保文件规范性、科学性和兼容性。GMP文件审核职责对照表见表1。

表1 GMP文件审核职责对照表Tab 1 GMP document audit responsibilities comparison table
4.3 强化培训管理,提升培训效果
依据GMP文件生命周期管理模型,在文件生效前,企业应对相关人员进行培训。首先,企业高层必须充分认识到培训的重要性,不得以各种理由取消或应付培训。只有每个员工都系统学习、掌握岗位相关文件的有关要求,理解规范操作的重要性,认识到个体行为的偏差对整个药品质量带来的危害,GMP才能得到正确贯彻。
其次,培训内容要有系统性、针对性。培训师应该用通俗易懂的语言将起草背景、相关专业知识、文件内容、执行要点、检查考核点以及执行文件时可能出现的问题、解决方法作系统而详细的讲解,帮助员工正确理解和使用文件,提高员工的质量意识和工作技能[6]。
再次,考核指标化,跟踪常态化。培训后应进行考核,与员工的待遇挂钩,以确保培训的效果[7]。要求真实填写并保存培训登记表、受训人员签名表、考核记录、培训效果评估表等资料。加强培训后跟踪,必要时对员工进行再培训,保存再培训的相关记录。
4.4 完善奖惩机制,加强文件执行力度
一套科学、系统、严谨、可行的文件系统,只有切实落实到实际工作中才能真正发挥作用,才能确保企业质量管理体系的有效性,促进药品生产过程科学化、规范化管理。针对当前许多企业存在的文件执行不到位的问题,笔者认为除了要强化培训外,还应建立一套完善的奖惩机制,将各部门、各岗位的工作职责细化,制订成具体、明确的评分项目,根据工作的落实完成情况进行评分;质管员、质量管理负责人、总经理等加强对实际生产操作过程的监督与纠正,并随时进行现场监督评分;每月评分与奖金挂钩,让员工树立起较强的质量意识和责任感,规范操作行为,并在日常工作中不断强化,上升为一种真正的习惯。其次,有效沟通,形成良好的企业文化。在文件执行过程中遇到困难或出现问题时,管理者与执行者应及时沟通,及时发现问题,并尽快解决,确保文件有效性和先进性,鼓励对GMP文件的创新改进。
4.5 科学存放,便于查阅
建立专门的GMP文件档案室,按照GMP文件的类型进行科学分类、排列、编号,建立档案存放位置索引,明确具体的存放位置,以便迅速存取档案。鉴于GMP对不同的文件提出的保存期限不同,建议企业建立“文件保管期限表”,列出不同文件的保存期限,按时处理,防止遗忘和差错。同时,定期检查档案保管情况,保证档案的安全、完整。
不少制药企业现有的电子文件管理方法较落后,需要升级改造或建立新的管理方法[8]。如建立电子文件流转跟踪登录管理,限制对电子文件的操作权限,确保不出现任意篡改或者未经授权进入等情形[9]。任何更改和删除记录应在权限范围内进行,其更改和删除原因和过程应在记录中得到体现。由于电子文件载体不稳定,应采用磁带、缩微胶卷、纸质副本或其他方法进行备份,防止信息损失。此外,建立电子文件检索目录,可与相对应的纸质文件进行关联,便于查找。
4.6 开展文件的变更控制
文件管理系统作为药品生产质量管理体系的一个基本要素,要始终满足药品生产质量管理体系的有效运行。所有受变更影响的文件都应该按实际情况进行评估和修订,并适时地进行控制。只有确保所有受影响的文件修订完成之后,质量管理负责人才能核准实施变更[10]。任何人可提出文件变更申请,由文件原审核人、批准人评价变更的必要性。在已变更文件生效时,应收回所有相对应的旧文件,保证使用文件为批准的现行文本。文件管理部门应及时完成因某个文件的变更而引起的其他相关文件的变更。文件的任何变更必须进行详细的记录,以便追踪检查。
5 结语
GMP文件生命周期管理模型是一项重要的理论创新和突破,其对指导和解释我国制药企业文件管理中的一些实践问题具有重要的现实意义。建议制药企业可依据该模型全面剖析本企业在文件管理中存在的问题,制订切实的整改措施和方案,提升质量管理水平,达到GMP认证要求。
[1]国家食品药品监督管理总局.2013年7月各省新修订药品GMP认证企业进度情况[EB/OL].(2013-08-15)[2013-08-17].http://www.sda.gov.cn/WS01/CL0896/83294.html.
[2]敖津京.电子文件时代文件生命周期理论的生命力[J].企业导报,2013(9):296.
[3]汪达.浅谈新版GMP文件系统编写[J].机电信息,2012(5):26.
[4]Kashiwagi D,Kashiwagi J.A new risk management model[J].J Risk Analysis Crisis Response,2012,2(4):237.
[5]张炜,任锐龙.对药品生产企业实施GMP的调查与思考[J].中国药事,2011,25(2):187.
[6]王小卫.浅谈新版GMP认证药品生产企业的培训管理[J].广州化工,2013,41(17):263
[7]梁毅.论药品生产企业GMP理念的建立[J].中国药房,2011,22(45):4225.
[8]Stamatiadis D,Scribner S.A robust methodological approach for replacing global electronic document management[J].Drug Inf J,2009,43(3):243.
[9]金朝阳,何蕾.纸质文件与电子文件管理的相互关系[J].兰台内外,2010(5):59.
[10]Bland P.Assessing the impact of changes on GMP systems and documents[J].J GXP Compliance,2005,9(4):19.
猜你喜欢
金桥(2022年5期)2022-08-24
现代仪器与医疗(2022年2期)2022-08-11
办公室业务(2020年18期)2020-09-29
家庭影院技术(2020年6期)2020-07-27
中国外汇(2019年13期)2019-10-10
劳动保护(2019年7期)2019-08-27
民用飞机设计与研究(2019年2期)2019-08-05
消费导刊(2018年10期)2018-08-20
中国医疗保险(2017年1期)2017-05-18
中国医疗保险(2017年1期)2017-05-18
