
气相色谱测定十滴水中樟脑和桉油精方法的改进
2014-11-25 09:55刘元媛于世海胡佳何涛
锦州医科大学学报 2014年5期
刘元媛,于世海,胡佳,何涛
(1.辽宁医学院,辽宁 锦州 121001;2.辽宁康辰药业有限公司,辽宁 丹东 118301;3.锦州市药品检验所,辽宁 锦州 121000)
气相色谱法是测定药物挥发性成分的主要分析手段,在药物分析方法中占有重要地位。我校已开展了近十年的气相色谱分析实验,但方法陈旧,并且实验用药已经退出市场,需要选择新的实验方法及药物来满足学生实验的教学需要。本课题组进行了多项研究,先后尝试了藿香正气水、复方丹参片、十滴水中挥发性成分的测定研究,并摸索了在不同气相色谱柱上的分离效果[1],最终确立了采用SE-54 毛细管柱测定十滴水中樟脑和桉油精含量的方法。该方法简便,精密度、重复性高,阴性液无干扰,可应用于学生实验教学。
1 仪器与试药
日本岛津GC-2010 气相色谱仪;AR2140 电子分析天平。樟脑对照品(北京北纳创联生物技术研究院,批号76-22-2);桉油精(中国药品生物制品检定所,批号:110788-201105);十滴水样品(广西慧宝源制药有限公司,批号 130325;北京海德润制药有限公司,批号130403);乙纯(分析纯),水为重蒸水。
2 实验条件
2.1 色谱条件的选择[1-5]
色谱柱:SE-54 石英毛细管柱(30 m ×0.25 mm ×0.25 μm);程序升温(70 ℃~160 ℃,升温速率为8 ℃/min);汽化室温度230 ℃;检测器:FID,温度240 ℃;载气:高纯氮气 (压力为120 kP);分流进样 (分流比为40∶1);内标物为环己酮,进样量1 μL。
2.2 内标溶液制备
精密量取环己酮适量,加70% 乙醇制成每1 mL 含10.481 mg 的溶液,作为内标溶液。
2.3 对照品溶液制备
精密称取樟脑、桉油精对照品适量,加70%乙醇制成每1 mL 含樟脑0.756 mg、含桉油精0.831 mg 的溶液,作为对照品溶液。
2.4 供试品溶液制备
取十滴水样品10 瓶,混匀,精密量取2.0 mL,置25 mL 量瓶中,精密加入内标液2.0 mL,加70%乙醇稀释至刻度,摇匀,即得。
2.5 阴性液的制备
按照处方工艺,制备缺樟脑和桉油精的样品,并按照供试品溶液的制备方法处理样品,得到樟脑和桉油精的阴性液。
2.6 方法学考察
2.6.1 系统适用性实验 分别取对照品溶液及供试品溶液各1 μL,按“2.1”项下色谱条件注入气相色谱仪,记录气相色谱峰。结果显示,溶剂峰、环己酮峰、桉油精峰、樟脑峰均完全分离,理论塔板数按樟脑计不低于50 000。
2.6.2 线性关系考察 精密取樟脑、桉油精对照品适量,加70% 乙醇制成每1 mL 含樟脑5.671 1 mg、含桉油精7.479 0 mg 的溶液,分别精密量取该溶液1.0、0.8、0.6、0.4、0.2 mL 置5 mL 量瓶中,分别精密加入内标液0.4 mL,加70%稀释至刻度,摇匀。精密吸取各溶液1 μL,按“2.1”项下色谱条件注入气相色谱仪,记录色谱峰。以对照品浓度为横坐标,对照品与内标物峰面积之比为纵坐标,绘制标准曲线,得到回归方程分别为:Y=2.091X-0.002 9,r=0.999 8,Y=1.201 9X-0.183 1,r=0.999 7。结果显示,樟脑在0.226 8~1.134 2 mg·mL-1范围内线性良好,桉油精在0.299 2~1.495 8 mg·mL-1范围内线性良好。
2.6.3 精密度试验 精密吸取对照品溶液1 μL,按“2.1”项下色谱条件注入气相色谱仪,重复进样5 次,测定樟脑、桉油精峰面积与内标物的峰面积比值,求得相对标准偏差RSD 分别为0.6%、0.5%。
2.6.4 稳定性试验 精密吸取同一供试品溶液1 μL,分别于0,2,4,6,8 h 按“2.1”项下色谱条件注入气相色谱仪,记录色谱图,求得樟脑、桉油精峰面积与内标物的峰面积比值的标准偏差RSD 分别为0.7%、0.8%,结果表明樟脑、桉油精在8 h 内稳定性良好。
2.6.5 重复性试验 取同一批样品5 份,按照“供试品溶液制备”项下方法制备供试品溶液,分别吸取各供试品溶液1 μL,按“2.1”项下色谱条件注入气相色谱仪,记录色谱图,求得樟脑、桉油精峰面积与内标物峰面积比值的标准偏差RSD 分别为1.8%、2.1%。
2.6.6 加样回收率试验 精密量取5 份含量已知的同一批号样品(樟脑的含量为22.97 mg·mL-1、桉油精含量为7.86 mg·mL-1)溶液各0.2 mL,置于10 mL 量瓶中,分别精密加入樟脑对照品溶液 (浓度为1.374 mg·mL-1)3.0 mL、加入桉油精对照品溶液 (浓度为2.493 mg·mL-1)0.3 mL、加入内标液0.2 mL,加70%乙醇稀释至刻度,摇匀,分别精密吸取各溶液1 μL,按“2.1”项下色谱条件注入气相色谱仪,记录色谱图,结求得樟脑的平均加样回收率为98.3%,RSD 为1.9%,桉油精的平均加样回收率为97.6%,RSD 为2.1%。
2.7 样品测定 取各批号样品,按照“供试品溶液制备”项下方法制备供试品溶液,分别吸取各供试品溶液1 μL,按“2.1”项下色谱条件注入气相色谱仪,记录色谱图,求得樟脑、桉油精的含量结果见表1。

表1 十滴水中樟脑、桉油精含量测定结果
3 讨 论
结合目前气相色谱应用的实践情况,本文选用了程序升温法同时测定两种物质的含量,使教学与实践紧密结合,若考虑实验成本也可以采用程序升温法测定其中一个成分的含量。实验用药选用十滴水,价格便宜,配制样品采用70%乙醇为溶剂,操作方便、无污染。
本实验分别考察了DB-WAX 毛细管色谱柱及SE-54毛细管气相色谱柱,结果显示在DB-WAX 毛细管色谱柱上分离效果不佳,而在SE-54 毛细管气相色谱柱上获得了良好的分离效果,因此选用SE-54 为测定用柱。
本实验分别考察了以环己酮、水杨酸甲酯为内标物,结果显示样品阴性液在水杨酸甲酯出峰处有干扰,因此选用环己酮为内标物。本方法采用的色谱条件与中国药典中十滴水的测定条件不同,但对样品的含量检测结果准确度较高,结果符合药典的相关要求,对科研教学具有一定的参考意义。
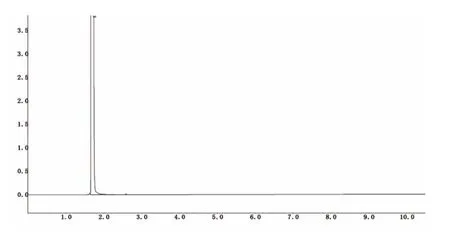
图1 溶剂乙醇气相色谱图
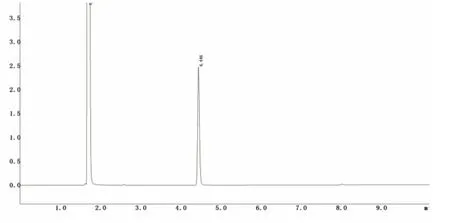
图2 环己酮气相色谱图
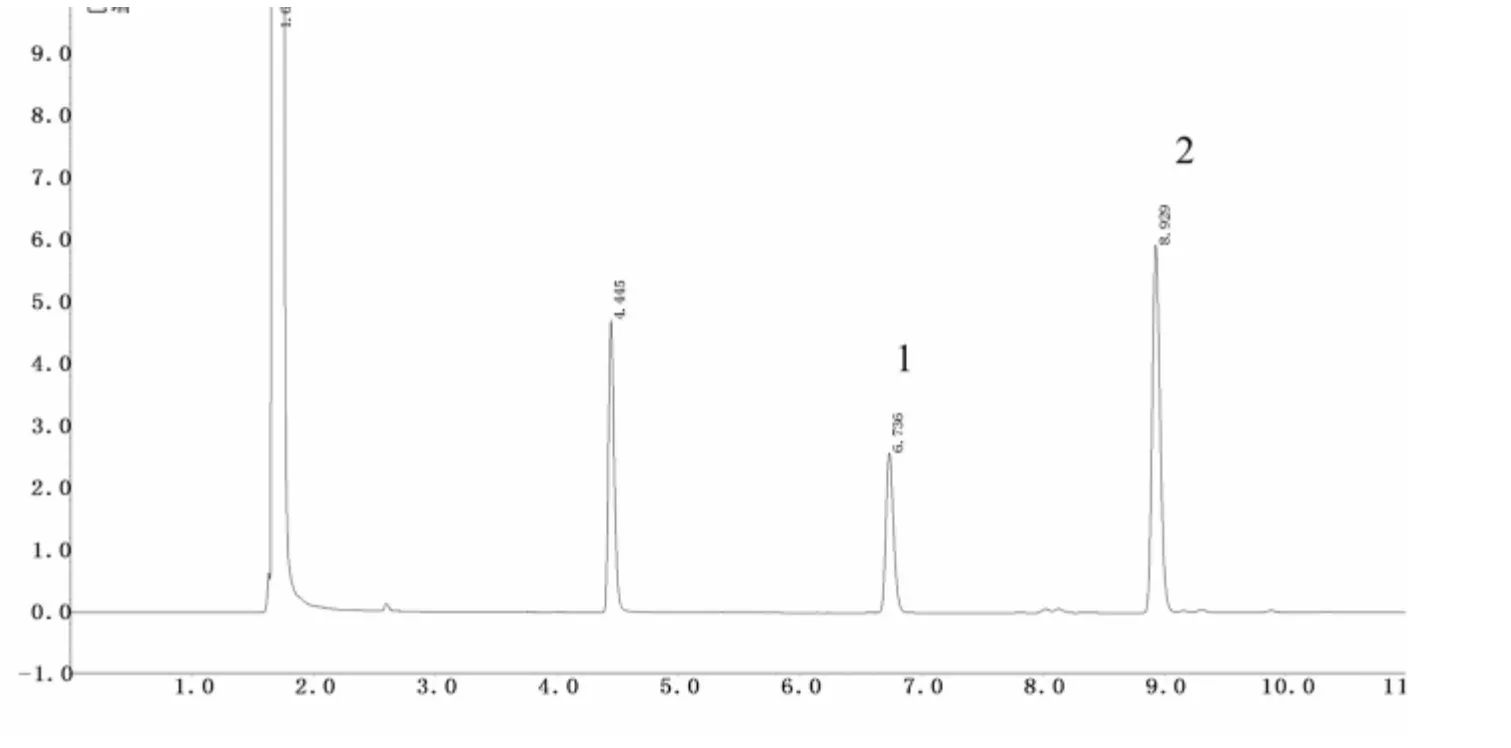
图3 樟脑、桉油精对照品气相色谱图
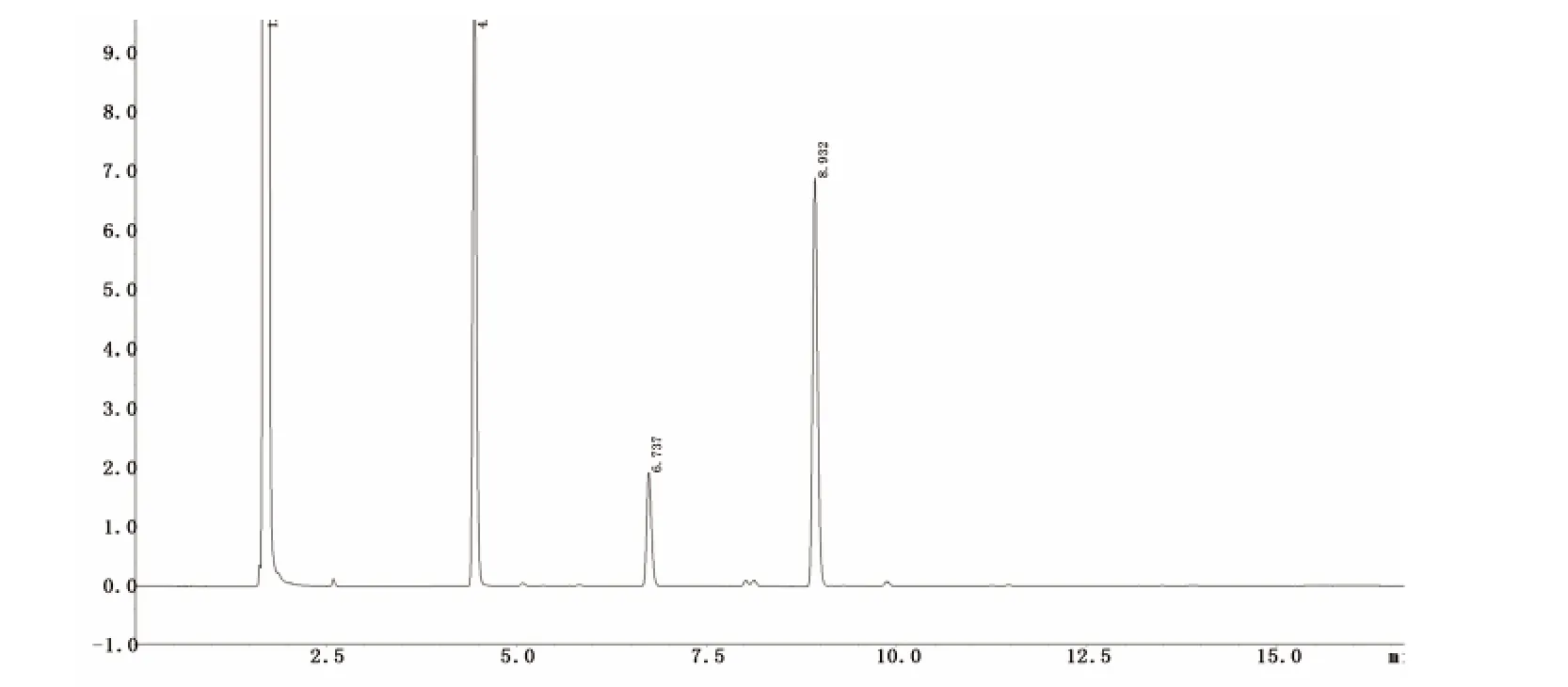
图4 十滴水样品气相色谱图
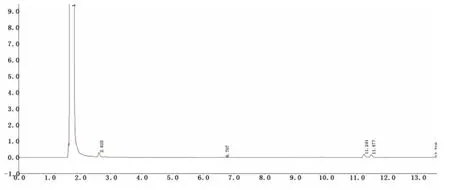
图5 樟脑桉油精阴性液气相色谱图
[1]卢华,张国宏,蒋受军,等.气相色谱法测定十滴水中樟脑和桉油精含量的研究[J].中成药,2008,30 (7):997-999.
[2]中国药典2010 年版一部[S].2010:301.
[3]王术玲,曾元儿,张迪,等.《中药制剂分析》气相色谱法含量测定的实验教学改进[J].中国中医药现代远程教育,2010,8 (23):43-44.
[4]王燕,张雅慧,格桑索朗,等.毛细管气相色谱内标法同时测定藏木香中土木香内酯、异土木香内酯的含量[J].药物分析杂志,2009,29 (12):2058-2060.
[5]靳然,于密密,赵百孝,等.气相色谱法测定艾叶4 个挥发性成分的含量[J].药物分析杂志,2013,33 (6):1033-1036.
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02
中国特种设备安全(2021年12期)2021-04-26
中华养生保健(2020年10期)2021-01-18
石油化工自动化(2018年5期)2018-11-14
中成药(2018年6期)2018-07-11
现代工业经济和信息化(2016年4期)2016-05-17
百姓生活(2016年3期)2016-03-25
中国粮油学报(2016年5期)2016-01-23
河北医药(2015年21期)2015-12-08
