
多活性中心催化剂催化丙烯聚合的均匀球Monte-Carlo模型
2014-11-20 03:49:14洪东峰隋述会吴文辉
合成树脂及塑料 2014年4期
洪东峰,隋述会,吴文辉*
(1. 北京理工大学材料学院,北京市 100081;2. 中科合成油技术有限公司,北京市 101407)
目前,约90%的聚丙烯(PP)采用Ziegler-Natta(Z-N)催化剂合成[1]。人们通过优化聚合工艺参数使催化剂性能尽可能发挥,从而提高PP性能。PP生产中,在催化剂作用下,丙烯以催化剂颗粒为核心参与聚合,从最初的10~200 μm的小粒径催化剂核心逐渐生长为粒径1 000~5 000 μm的PP颗粒[2-3]。MgCl2负载型多活性中心Z-N(MgCl2-Z-N)催化剂的催化动力学十分复杂,在丙烯聚合过程中起支配作用,同时丙烯在催化剂颗粒中的扩散也具有重要作用[4]。催化机理模型通常分为化学模型(动力学控制)和物理模型(扩散控制)。化学模型得益于数学的不断发展,如连续变量逼近法、矩阵法、Galerkin h-p有限元法等,近年来Monte-Carlo方法的应用越来越多,且更注重利用微分方程研究催化动力学[5],甚至开发了动态Monte-Carlo模型,描述聚合时间对聚烯烃链长分布的影响[6],但这些方法通常不考虑聚合过程中的传质和/或传热[6-7]。物理模型主要考虑聚合过程中的传质和/或传热等扩散作用[3,8],扩散理论认为:在径向上,催化剂裂片分布在不同浓度的单体和链转移剂(主要是H2)中,得到的聚合物链段基本相同,但由于空间位阻效应而形成不同的立构分布。物理模型的应用并不普遍。事实上,丙烯聚合过程中的化学作用和物理作用是同时存在的,且两者相互影响,Sun Jianzhong等[8-9]利用多层模型对以上因素进行了研究,使丙烯聚合催化模型的建立较之前有很大进步。研究表明,Z-N催化剂在MgCl2上的负载并非呈层状,而是均匀分布[5],因此,多层模型与MgCl2-Z-N催化剂的催化过程尚有不同。
本研究应用Monte-Carlo方法对丙烯聚合均匀球模型进行积分处理,建立均匀球Monte-Carlo模型(UBMCM)。采用中国石油化工股份有限公司开发的第四代多活性中心催化剂DQ-Ⅲ,实验数据来自工业PP环管装置。
1 模型建立
Zacca等[2]首先提出Z-N催化剂的动力学机理:不同氧化态的Ti具有不同的催化性能,从而使催化剂具有多种活性中心。通过解析PP的凝胶渗透色谱(GPC)曲线,确定DQ-Ⅲ催化剂具有5个活性中心,决定系数为0.999 98,决定系数越接近1,解析结果(见图1)越可靠。
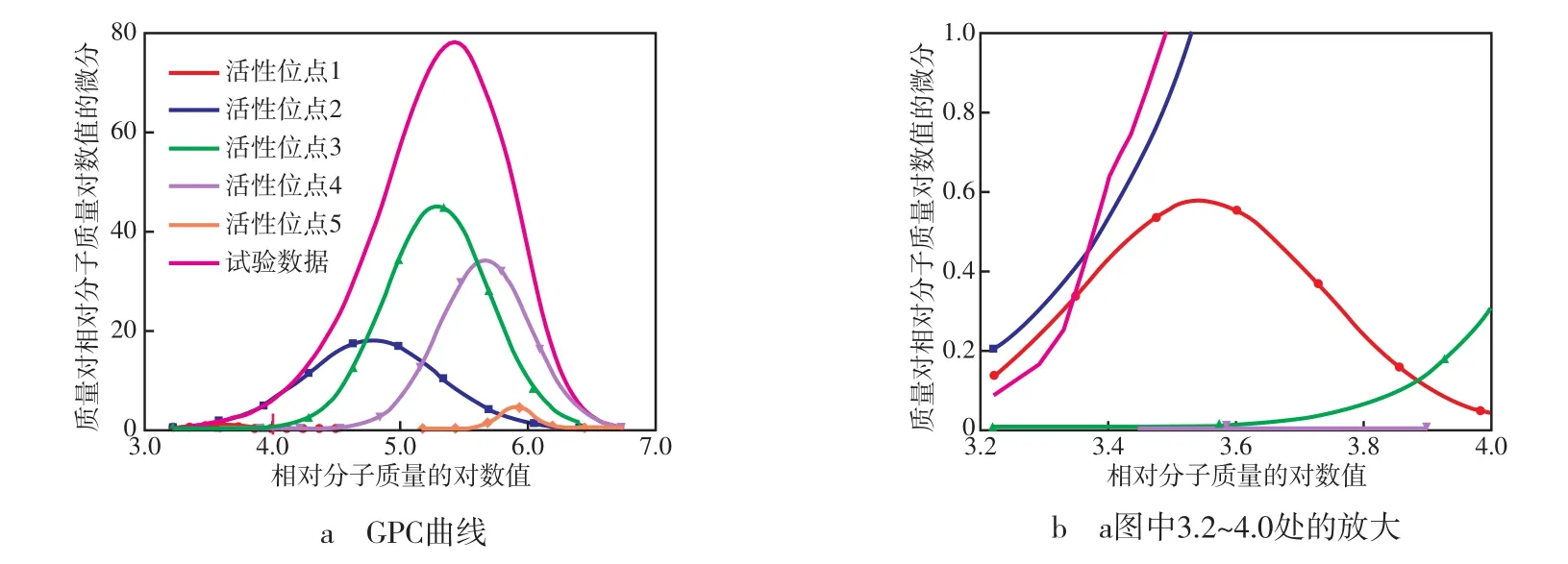
图1 DQ-Ⅲ催化剂的活性中心解析结果Fig.1 Analytical results of the active sites in DQ-Ⅲ catalyst
针对第四代MgCl2-Z-N催化剂引发的丙烯聚合,提出以下假设[2-3]:1)活性中心在催化剂内的分布,以及单体、催化剂、H2等在反应器内的分布均匀;2)加入反应器中的催化剂颗粒粒径相同;3)催化剂和/或PP颗粒内/间的热交换可以忽略;4)聚合初期,催化剂颗粒的破裂在瞬间完成。
聚合初始阶段,催化剂破裂成若干小颗粒且能保持球形,丙烯单体通过扩散进入催化剂内部与活性中心接触并发生反应,聚合过程中,催化剂作为丙烯聚合的模板和微反应器,以催化剂为核心,经历链引发、链增长和链转移,得到一个PP颗粒[9]。催化动力学模型由活性中心活化、链引发、链增长、链转移、活性中心转移、活性中心失活等6个过程及每个过程的速率常数来描述。
UBMCM中,丙烯分子透过催化剂外壳,在扩散作用下进入催化剂内部,遵循Fick扩散定律。丙烯单体在催化剂颗粒中的扩散见图2。
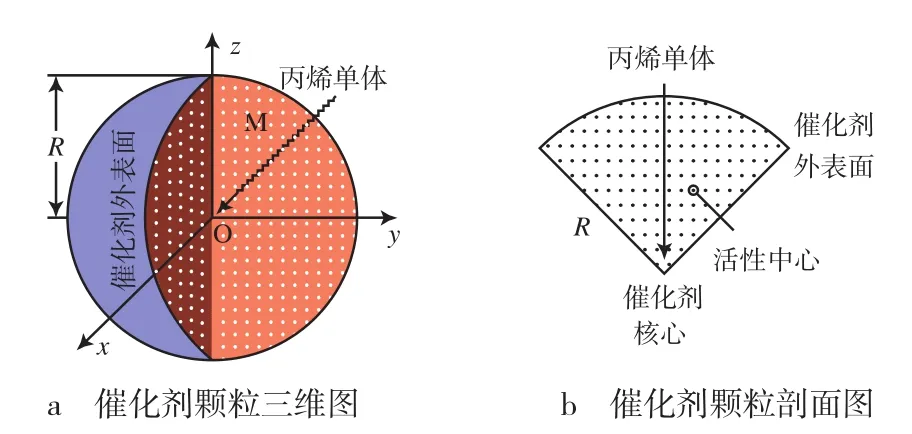
图2 丙烯单体在催化剂颗粒中的扩散Fig.2 Diffusion of the propylene monomer in catalyst particles
丙烯的浓度梯度计算见式(1)~式(2)。
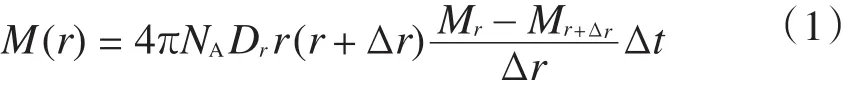
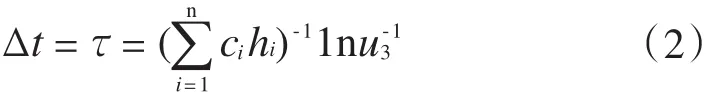
式中:M(r)表示丙烯的浓度梯度;NA是Avogadro常数;Dr是r处的扩散系数,r是活性中心与球心间的距离;Δr为半径的变量,Δr<0;Mr和Mr+Δr分别是r和r+Δr处的丙烯浓度;τ和Δt是设定的时间间隔;ci是stochastic速率常数;hi是参与第i种基元反应的分子数;n是总基元反应数;u3是随机数。
单体丙烯自催化剂外向其核心扩散,当扩散至[r, r+Δr]时,催化剂颗粒的体积按式(3)计算。
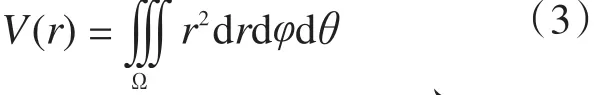
半径的积分区间是[r, r+Δr],φ和θ的积分区间均为[0, 2π],用式(4)~式(6)表述。


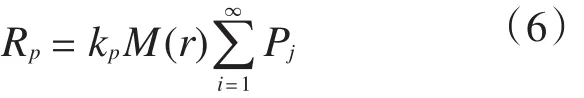
式中:kp是聚合反应速率常数;Pj是第j种反应的几率;Rp是聚合反应速率;ρp是催化剂/PP复合颗粒的密度;Mw是PP的相对分子质量;Vr,Vr+Δr分别是时间(t)和t+Δt时复合颗粒的体积。
随着聚合的进行,催化剂/PP颗粒的半径和体积逐渐增加,但其密度保持不变,故半径或体积的增加与参加反应的丙烯量有关。
UBMCM的算法流程见图3。计算在Matlab 2012b软件上进行。
2 结果与讨论
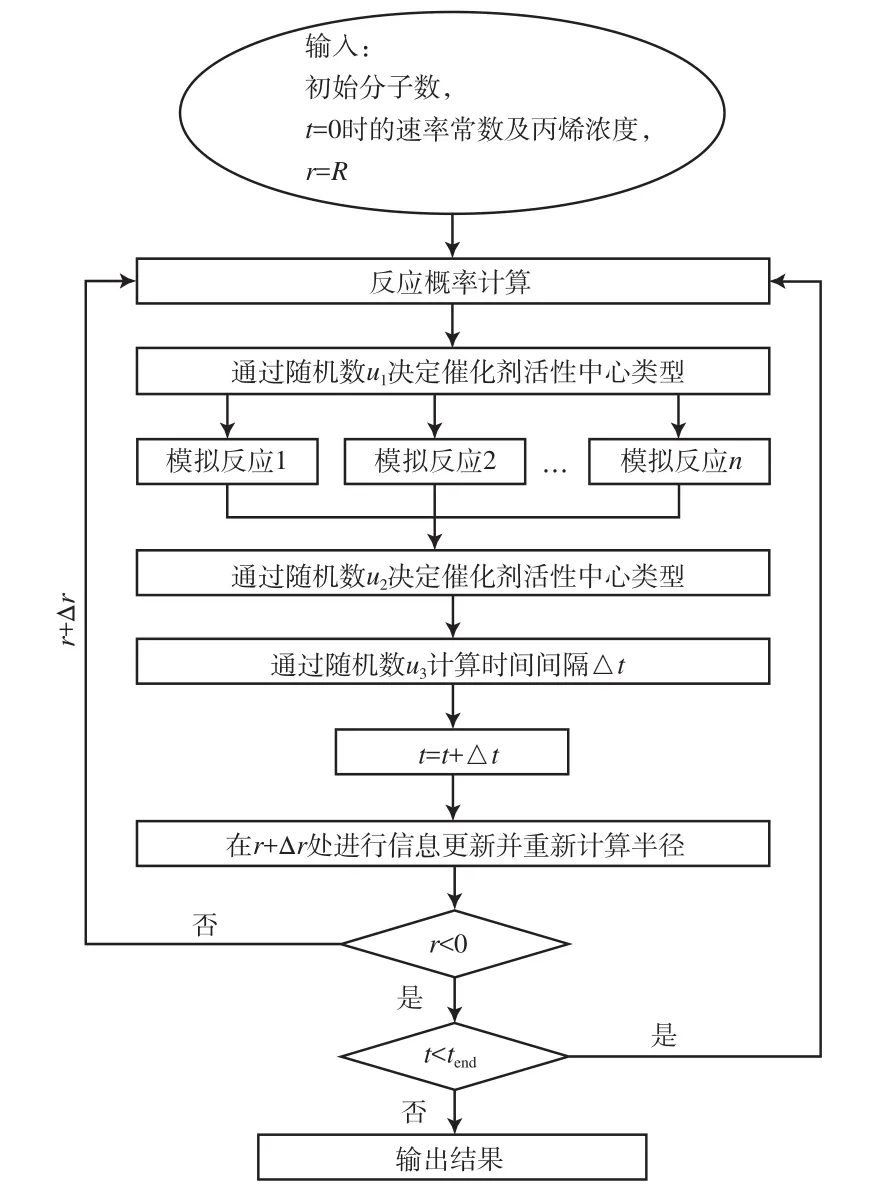
图3 UBMCM的算法流程Fig.3 Algorithm flowchart of the UBMCM model
采用UBMCM模拟丙烯聚合的反应动力学,温度为70 ℃,分析了DQ-Ⅲ催化剂的活性,PP的Mw、相对分子质量分布(Mw/Mn)和粒径分布,并与Zacca等[2,10]使用的Monte-Carlo模型(MCM)和Wang Wei等[9]使用的多层Monte-Carlo模型(MLMCM) 的模拟结果进行比较。
2.1 催化剂活性
用UBMCM模拟多活性中心Z-N催化剂催化丙烯聚合时,预测结果与实验结果间的相关系数为0.994 1,而用MCM和MLMCM时的相关系数分别为0.992 3和0.992 5[2,9]。3种模型中,UBMCM曲线与实验点吻合最好,说明UBMCM对该现象的描述较好。这是因为随着聚合的进行,催化剂颗粒内部的单体浓度梯度变小,Fick扩散的动力降低。用UBMCM预测的结果与实验值吻合度高于用MCM和MLMCM,是因为UBMCM考虑了扩散对丙烯聚合的影响,模型更精确。从图4可以看出:聚合前期,扩散阻力对聚合不利,而后期有利于聚合,聚合过程可分为3个阶段。第一阶段(0~1.0 h)为扩散控制,由于扩散阻力,催化剂内部及活性中心周围单体较少,催化剂浓度低,扩散过程非常缓慢,且占主导地位,催化剂活性较低,但随着单体向催化剂内部扩散,催化剂颗粒内丙烯浓度增加,催化剂活性增加较快(曲线斜率大)。第二阶段(1.0~4.0 h)为反应控制,单体开始在催化剂活性中心周围积累,单体浓度增加,同时扩散逐渐趋于平衡,催化剂活性增加趋势减缓(曲线斜率减小)。第三阶段(4.0~6.0 h)为稳态阶段,由于失活反应,故单体和活性中心的浓度均降低,催化剂活性不再有明显变化。

图4 多活性中心催化剂活性与聚合时间的关系Fig.4 Relationship between the multi-active-site catalyst and the polymerization time
2.2 PP的Mw
从图5可以看出:相对于MCM与MLMCM,用UBMCM模拟时由于考虑了扩散的影响,对Mw的预测值与实验值吻合度更高,对丙烯聚合的描述更精确。随着聚合的进行,Mw逐渐增加,直至接近最大值,其中,用UBMCM的预测曲线比用MLMCM的平滑。

图5 PP的Mw与聚合时间的关系Fig.5 Relationship between the relative molecular mass of PP and the polymerization time
2.3 PP的Mw/Mn
从图6可以看出:对于多活性中心催化剂引发丙烯聚合,聚合3.5~4.0 h时,PP的Mw/Mn逐渐趋于恒定,约为4.4。造成Mw/Mn变宽的原因有两个:催化剂的多活性中心性质和扩散作用。聚合过程中,活性中心由Ti3+转换成Ti2+,Ti2+与丙烯之间的反应不活跃是造成PP的Mw/Mn变宽或其曲线出现双峰的主要原因。用UBMCM预测的Mw/Mn与工业或实验数据的吻合度最高,MLMCM次之,MCM最低,因此,UBMCM在精确模拟丙烯聚合和预测产品性能方面有优势。
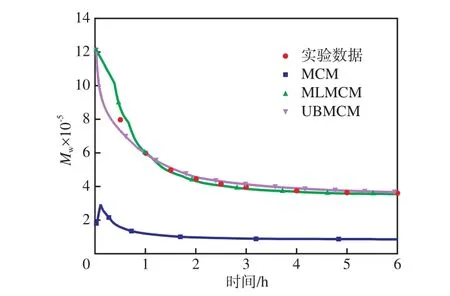
图6 PP的Mw/Mn与聚合时间的关系Fig.6 Relationship between Mw/Mn of PP and the polymerization time
2.4 PP的粒径
聚合过程中,单/多活性中心催化剂催化生成的PP的粒径变化趋势基本相同。从图7看出:扩散控制阶段,即第一阶段,催化剂活性中心周围的单体浓度低,PP粒径增速较慢;反应控制阶段,即第二阶段,在第一阶段单体浓度梯度作用下,催化剂内部单体浓度增加,PP粒径增速较快;稳态阶段,即第三阶段,PP粒径增速减小并趋于稳定。
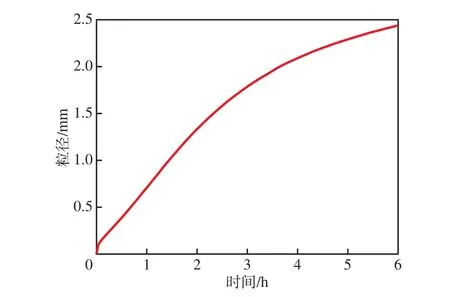
图7 PP粒径与聚合时间的关系Fig.7 Relationship between the particle size of PP and the polymerization time
3 结论
a)根据UBMCM模拟结果,聚合反应动力学和颗粒间/内的扩散效应对催化剂的催化性能和PP性能均有重要影响。
b)丙烯聚合过程可分为扩散控制、反应控制和稳态3个阶段。
c)UBMCM考虑到扩散阻力,对多活性中心催化剂催化丙烯均聚合过程的描述更精确,对PP性能的预测更准确。
[1] Zhang Hexin, Lee Y-J, Park J-R, et al. Control of molecular weight distribution for polypropylene obtained by a commercial Ziegler-Natta catalyst: effect of a cocatalyst and hydrogen[J].Journal of Applied Polymer Science, 2011, 120(1): 101-108.
[2] Zacca J J, Debling J A, Ray W H. Reactor residence time distribution effects on the multistage polymerization of olefins—I. Basic principles and illustrative examples, polypropylene[J].Chemical Engineering Science, 1996, 51(21): 4859-4886.
[3] McKenna T F, Soares J B P. Single particle modelling for olefin polymerization on supported catalysts: a review and proposals for future developments[J]. Chemical Engineering Science, 2001,56(13): 3931-3949.
[4] Cifra P, Bleha T. Stretching of self-interacting wormlike macromolecules[J]. Polymer, 2007, 48(8): 2444-2452.
[5] Jiang Xue, Wang Hang, Tian Xiuzhi, et al. Effects of doping LiCl into MgCl2-supported Ziegler-Natta catalyst on the molecular weight distribution and isotacticity of polypropylene[J].Industrial & Engineering Chemistry Research, 2011, 50(1):259-266.
[6] Simon L C, Soares J B P. Monte Carlo simulation of long-chain branched polyolefins made with dual catalysts: a classification of chain structures in topological branching families[J]. Industrial &Engineering Chemistry Research, 2005, 44(8): 2461-2468.
[7] Luo Zhenghong, Wang Wei, Su Peilin. Modeling of the propylene polymerization catalyzed by single-/multi-active site catalyst: a Monte Carlo study[J]. Journal of Applied Polymer Science, 2008, 110(6): 3360-3367.
[8] Sun Jianzhong, Eberstein C, Reichert K H. Particle growth modeling of gas phase polymerization of butadiene[J]. Journal of Applied Polymer Science, 1997, 64(2): 203-212.
[9] Wang Wei, Zheng Zuwei, Luo Zhenghong. Coupled-singleparticle and Monte Carlo model for propylene polymerization[J].Journal of Applied Polymer Science, 2011, 119(1): 352-362.
[10] Di Drusco G, Rinaldi R. Polypropylene-process selection criteria[J]. Hydrocarbon Process, 1984, 63(11): 113-117.
猜你喜欢
扬子江诗刊(2023年3期)2023-05-06 10:40:14
大众文艺(2022年16期)2022-09-07 03:08:04
中国食用菌(2021年10期)2021-11-04 06:23:20
农药科学与管理(2019年5期)2019-08-13 00:48:02
中国军转民(2017年7期)2017-12-19 13:30:00
北京航空航天大学学报(2017年2期)2017-11-24 05:24:51
当代化工研究(2016年7期)2016-03-20 16:21:55
大连工业大学学报(2015年4期)2015-12-11 04:06:50
天津科技大学学报(2015年4期)2015-04-16 04:55:09
中国卫生(2014年10期)2014-11-12 13:10:24
