
甲型H1N1流感病毒三维空间结构预测
2014-11-09 00:44靳佩轩
食品与生物技术学报 2014年5期
靳佩轩, 高 洁
(江南大学 理学院,江苏 无锡 214122)
流感病毒具有多样的变异性,历史上每一次流感大流行多是由流感病毒新亚型和以往出现过的亚型的再次出现,人类绝大多数对其缺乏相应的免疫力,并且现有的流感疫苗起不到有效作用,从而造成流感病毒在人群中快速广泛的传播,最终出现流感大流行[1-2]。面对流感病毒表面抗原蛋白结构复杂多变的性状和变异的突发性,研究流感病毒蛋白的三维空间结构显得特别重要。
近年来,关于蛋白质三维空间结构的研究已有不少。1989年,Dill and Lau[3]建立蛋白质空间结构的HP模型,忽视侧链的影响,将氨基酸分为亲水性(P)和疏水性(H)两类,从而使氨基酸序列抽象成一个二进制的序列,来构建一个存在疏水核的二维或三维蛋白质结构模型。2006年,陈凤飞[4]又衍生出关于HP模型的修正模型,即三角化的HP格点模型。2004年,Custodio等[5]提出用遗传算法去优化三维蛋白质HP格点模型,得到结构最优的三维结构。2010年,Vincent等[6]用从头计算的思想,利用分子动力学方法深入研究蛋白质三维空间结构折叠的原理,为从头计算建立三维空间模型进行相关因素分析。2011年,Ivan Dotu等[7]提到将格点模型和非格点模型用 LNS(Large Neighborhood Search)去找到HP模型的自然态。2011年,Islam[8]对蛋白质结构的三维HP格点模型用MA(memetic algorithm)搜索算法进行优化。2013年,张玲等[9]运用ARFIMA模型对甲型H1N1流感病毒的HA蛋白质的序列进行预测分析。2009年,Manabu Igarashi等[10]运用已经很成熟的比较建模的方法对2009年甲型H1N1流感病毒的结构进行了模建分析。
作者基于蛋白质空间结构的HP模型,将蛋白质结构中氨基酸种类分成两类,构建了甲型H1N1流感病毒蛋白质空间结构的3DHP模型,利用改良的遗传算法找到最小自由能结构,从而预测了甲型H1N1流感病毒蛋白质三维空间结构,并在PDB数据库中取了8条已测得的甲型H1N1流感病毒的蛋白质空间结构中心碳原子坐标数据,将甲型流感病毒蛋白的空间坐标转换为距离矩阵量化表示,对预测结构与实际测得的结构进行比较,通过相关性分析和显著性检验,表明预测结构与已知结构存在高度的一致性。
1 材料与方法
1.1 基于蛋白质空间结构的3DHP模型
将氨基酸按亲疏水性分类,即将20种氨基酸分两类: 疏水性 H={A,I,L,M,F,P,W,V}, 亲水性P={N,C,Q,G,S,T,Y,D,E,R,H,K}, 又令:H=1 和P=0,蛋白质的氨基酸序列即可转换为由0和1组成的序列。由蛋白质三维空间结构产生的主要驱动力是氨基酸的疏水效应[11],则在蛋白质结构中疏水残基间相互作用在蛋白质的中心形成一个疏水核,亲水残基包围在这个核的外面形成了一个稳定的蛋白质空间结构。
HP格点模型在三维空间中的折叠简称3DHP模型,将氨基酸看作一个节点,定义在模型中各节点间最小距离为单位1,在折叠过程中各节点位置不能重叠,每个节点的折叠方向在空间中有六个,即在立体方格中每个氨基酸节点可分别进行90°的向上(u)、下(d)、左(l)、右(r)、前(f)和后(b)六个方向的折叠(见图1a)。由于每个节点不能重复,确定前一个节点的位置后,下一个节点最多有5个折叠方向(见图1b)。此模型中任意两个节点不重合,并忽略折叠中侧链的影响,虽将蛋白质的空间结构简化,但是整体的蛋白质空间结构骨架符合真实蛋白质结构的基本特征,能很好地模拟真实蛋白的折叠行为,且计算简单,有利于对比不同折叠搜索算法。

图1 3DHP结构中氨基酸节点的折叠方向Fig.1 Folding direction about amino acid nodes in the 3DHP structure
1.2 能量函数
1973年Anfinsen提出了蛋白质的天然构象对应其能量最低的结构这一热力学假说。为搜索到3DHP模型构建的能量最小的蛋白质三维空间结构,在此定义其能量函数。
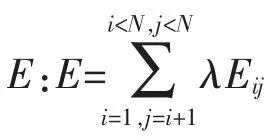

图2 序列PHPHHHPHHP的3DPH模型Fig.2Model of PHPHHHPHHP

图3 空间结构中空间相邻的节点间的能量表Fig.3 Energy value table about the space of the adjacent nodes in the structure
由图3可知,蛋白质空间结构中空间相邻的三种情况 1-1、0-1、0-0的能量值分别为 E11=-1,E01=0,E00=0,则具有最小自由能的蛋白质空间结构就是搜索得到空间相邻1-1个数最多的空间结构。
1.3 改良的遗传算法
改良的遗传(GA)算法主要引入局部优化策略,将GA算法和模拟退火(SA)算法结合,既有效地克服了SA算法求最优解耗时大的缺点,又有效地避免了GA算法因为其早熟收敛而得到非全局最优解的问题。
算法的基本步骤设计如下:1)由3DHP模型随机产生100个合法的蛋白质空间结构,计算其个体适应度,即每个空间结构的能量值;2)选择过程,采用随机选择思想在100个结构中随机选择两个结构作为算法优化目标,并计算各自能量值;3)单点交叉过程,针对步骤2中选出的两个结构,由前面可知这两个结构为同一个序列得到的不同的空间结构,则在这两个结构中分别随机选择其对应氨基酸序列上的同一个位置的氨基酸作为交叉位点,将两个结构中位于这个交叉位点后的结构进行互换得到两个新的空间结构,并读取两个新结构的节点坐标;4)变异过程,在进行交叉过程后形成的两个新的个体会发生结构中节点坐标重叠的现象,对重叠的节点进行变异操作,即依次改变交叉点后出现重叠的节点的折叠方向来保证新的个体为合法的蛋白质空间结构,并计算其各自的自由能;5)对得到的四个结构进行筛选,即从这四个结构中先选出一个自由能最低的结构作为下一个优化的目标,同时剔除自由能最高的一个结构,对剩下的两个结构根据其自由能大小,按照SA算法进行概率筛选;6)经过以上过程对蛋白质空间结构进行优化,重复步骤3~5直到产生自由能最低的空间结构,即视作自然状态下稳定的蛋白质空间结构。
2 甲型H1N1流感病毒的蛋白质空间结构
2.1 预测蛋白质空间结构
基于3DHP模型运用优化的遗传算法,分别以长度为13的HPPHPPHPHPPHP序列、长度为17的HHHHPPHHHHHHHPPPH序列、长度为20的PHHHHHHPHHHHPHHPHHPP序列和长度为21的PHPHPPHPHPPHPPHPHPPHP序列为例,并与文献[12]所得最低能量进行比较,比较结果见表1。图4~5给出长度分别为13和17的蛋白质序列的空间结构。

图4 长度为13的氨基酸序列3DHP模型(其能量E=-5)Fig.4 Sequence model about the length of the size for 13(the energy value E=-5)

图5 长度为17的氨基酸序列3DHP模型(其能量E=-9)Fig.5 Sequence model about the length of the size for 17(the energy value E=-5)

表1 3DHP预测模型最小能量比较Table 1 Minimum energy comparison on the 3DHP
2.2 预测甲型H1N1流感病毒蛋白质空间结构
选取8条甲型H1N1流感病毒HA蛋白质序列,编号分别为:1RUZ、1RUY、1RU7、3HTO、2WRH、3SM5、3UYW、4B7M (序列来源于pdb网站,网址:http://www.rcsb.org/pdb/home)。运用 3DHP 模型分别对 1RUZ的 H链、1RUY的 H链、1RU7的 H链、3HTO的 A链、2WRH的 H链、3SM5的 A链、3UYW的A链、4B7M的A链进行空间结构预测,获得每个氨基酸节点的空间坐标,并从PDB数据库中取相应的已知空间结构的中心碳原子的坐标,将其视为所在氨基酸的空间坐标,由此得到实际测得的蛋白质空间结构骨架,与预测所得的结构进行比较。
2.2.1 构建距离矩阵 对结构的比较采用将蛋白质的空间坐标转换为距离矩阵形式量化表示的方法。对长为n的序列,其中氨基酸节点的坐标分别为(x1,y1,z1),…,(xn,yn,zn),i=1,2,…,n。
定义距离矩阵A:

其中αij为在矩阵A的第i行j列的元素。
由此建立关于两个蛋白质空间结构的距离矩阵A中第i行、第j列的元素αij表示空间结构中第i个氨基酸节点到第j个氨基酸节点间的欧氏距离。即得到由模型预测所得结构和实验室测得结构的两个距离矩阵。
2.2.2 相关性分析 定义两个距离矩阵A和B的相关系数:

相关系数值越接近1,检验效果越显著,表示两距离矩阵越相似,即对应的两结构越相近,以此评价建立模型的建模效果。
取 1RUZ的 H链、1RUY的 H链、1RU7的 H链、3HTO的A链、2WRH的H链、3SM5的 A链、3UYW的A链、4B7M的A链的前20个氨基酸为例进行分析,运用3DHP模型分别对8条氨基酸链进行空间结构预测获得氨基酸节点空间坐标,并在PDB数据库中获取相应结构的坐标。
以 1RUZ的 H链 前 20个 氨 基 酸ATNADTICIGYHANNSTDTV为例,其在PDB数据库中的坐标见表2。运用3DHP模型分别对1RUZ的H链前20个氨基酸序列进行空间结构预测得到其空间结构,见图6,对应的氨基酸节点坐标见表3。

表2 1RUZ的H链在PDB中中心碳原子Cα的坐标(Aº)Table 2 Coordinates of center carbon atomsabout 1RUZ-H chain in the PDB
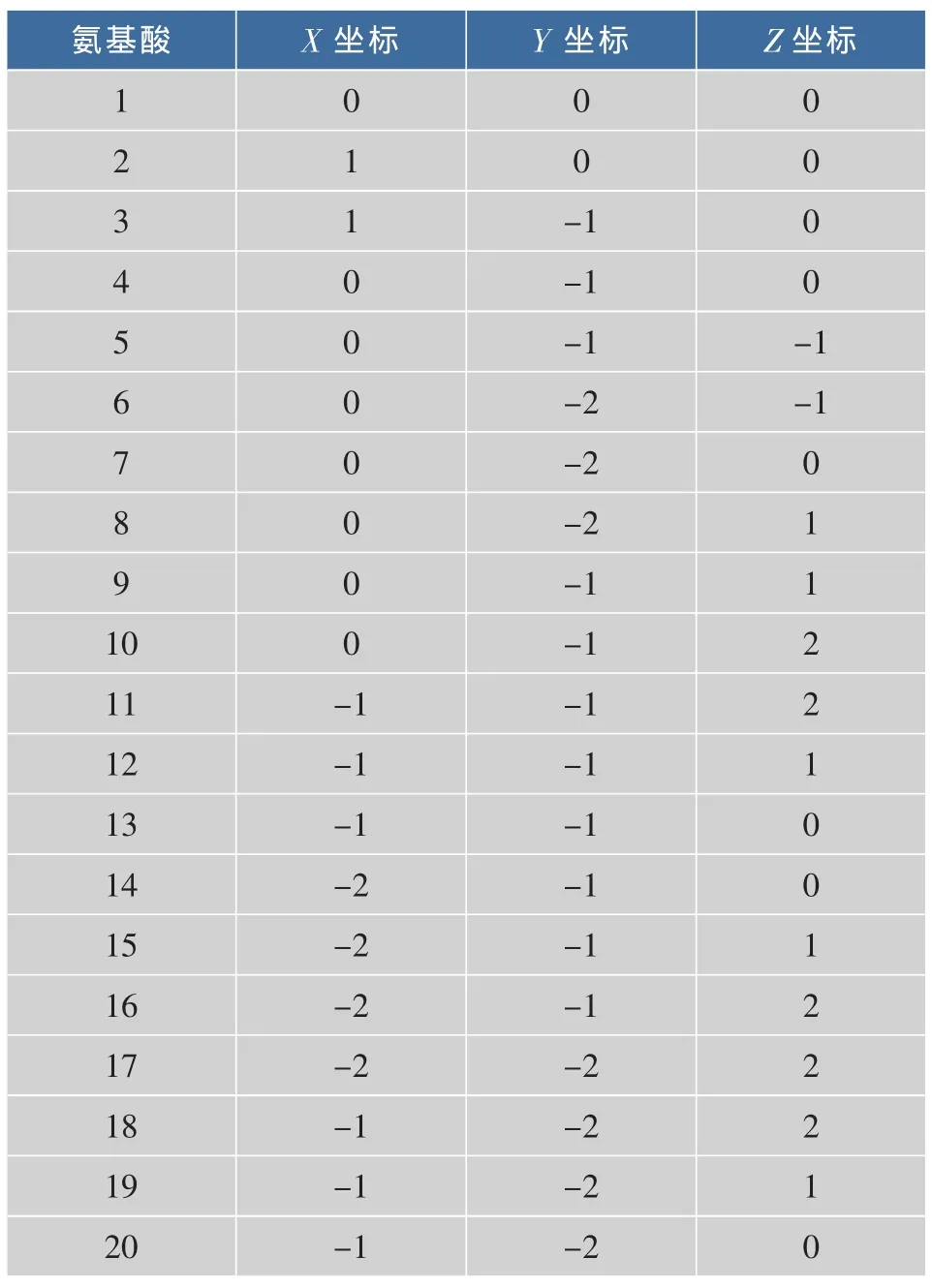
表3 1RUZ的H链模型氨基酸节点坐标Table 3 Node coordinates of amino acids on the model of 1RUZ-H chain

图6 1RUZ的H链结构预测模型Fig.6 Prediction model about 1RUZ-H chain structure
根据距离矩阵定义,对1RUZ的H链通过3DHP模型所得结构和PDB数据库提供结构分别构造距离矩阵进行相关性分析。求得两个距离矩阵的相关系数,P值为,可知这两个距离矩阵高度相关,即预测结构与已知结构存在高度的一致性。对另外7条:1RUY的H链、1RU7的H链、3HTO的A链、2WRH的H链、3SM5的A链、3UYW的 A链、4B7M的A链进行结构模建后空间结构坐标的距离矩阵相关性分析见表4,可知预测结构与已知结构也存在高度的一致性。

表4 其余7条序列的r值、p值Table 4 r value and p value of the rest of the 7 amino acids sequence
3 结语
目前来看,对流感病毒的研究大部分都是针对其一级结构的分析,基于理论对流感病毒蛋白质空间结构的研究还很少,虽Manabu Igarashi等运用比较建模的方法对2009年甲型H1N1流感病毒的结构进行了模建分析[9],但还很少有运用从头计算来对流感病毒蛋白质空间结构进行模建预测。作者采用3DHP模型和改良的遗传算法获取最小能量的空间构象,预测甲型H1N1流感病毒蛋白质空间结构,直接由一级结构预测甲型H1N1流感病毒的蛋白质三维空间结构,得到其空间结构数据,与PDB数据库中的实际结构进行比较分析得到了很好的检验结果,这就提供了一种快速预测甲型H1N1流感病毒结构的方法。
[1]Layne S P,Monto A S,Taubenberger J K.Pandemic influenza :an inconvenient mutation[J].Science,2009,323(5921):1560.
[2]Tokuriki N,Tawfik D S.Stability effects of mutations and protein evolvability[J].Current Opinion in Structural Biology,2009,19(5):596-604.
[3]Lau K F,Dill K A.A lattice statistical mechanics model of the conformational and sequence spaces of proteins[J].Macromolecules,1989,22(10):3986-3997.
[4]陈凤飞.蛋白质结构预测的三角化模型和算法[D].武汉:华中科技大学,2006.
[5]Custódio F L,Barbosa H J C,Dardenne L E.Investigation of the three-dimensional lattice HP protein folding model using a genetic algorithm[J].Genetics and Molecular Biology,2004,27(4):611-615.
[6]Voelz V A,Bowman G R,Beauchamp K,et al.Molecular simulation of ab initio protein folding for a millisecond folder NTL9(1-39)[J].Journal of the American Chemical Society,2010,132(5):1526-1528.
[7]Dotu I,Cebrian M,Van Hentenryck P,et al.On lattice protein structure prediction revisited[J].Computational Biology and Bioinformatics,2011,8(6):1620-1632.
[8]Islam M,Chetty M.Clustered memetic algorithm with local heuristics for ab initio protein structure prediction[J].2013,17(4):558-576.
[9]张玲,高洁.甲型流感病毒HA蛋白质序列的预测[J].食品与生物技术学报,2013,32(8):828-831.ZHANG Ling,GAO Jie.Prediction for base of influenza virus A/HA protein sequence[J].Journal of Food Science Journal of Health Laboratory Technology,2013,32(8):828-831.(in Chinese)
[10]Igarashi M,Ito K,Yoshida R,et al.Predicting the antigenic structure of the pandemic(H1N1)2009 influenza virus hemagglutinin[J].PLoS One,2010,5(1):e8553.
[11]Dill K A,Bromberg S,Yue K,et al.Principles of protein folding—a perspective from simple exact models[J].Protein Science,1995,4(4):561-602.
[12]ZHOU Changjun,HOU Caixia,ZHANG Qiang,et al.Enhanced hybrid search algorithm for protein structure prediction using the 3D-HP lattice model[J].Journal of Molecular Modeling,2013,19(9):3883-3891.
猜你喜欢
阿来研究(2020年1期)2020-10-28
广东医科大学学报(2020年6期)2020-02-06
中国洗涤用品工业(2019年4期)2019-05-11
中成药(2018年1期)2018-02-02
现代检验医学杂志(2016年3期)2016-11-15
现代检验医学杂志(2016年1期)2016-11-12
新世纪水泥导报(2016年1期)2016-07-01
中央社会主义学院学报(2016年2期)2016-05-04
动物医学进展(2015年10期)2015-12-07
华南农业大学学报(2015年5期)2015-12-04

- 食品与生物技术学报的其它文章
- 超高压处理对橙囊胞香气成分的影响
- 会议信息
- 科技信息
- 酿酒废水资源化关键技术及其应用研究