
马铃薯SGT3基因表达及其启动子功能分析
2014-11-07 01:46崔同霞白江平魏桂民赵旭王蒂张金文
草业学报 2014年2期
崔同霞,白江平,魏桂民,赵旭,2,王蒂*,张金文*
(1.甘肃省作物遗传改良与种质创新重点实验室 甘肃省干旱生境作物学重点实验室 甘肃农业大学农学院, 甘肃 兰州730070;2.甘肃省农垦农业研究院,甘肃 武威733006)
糖苷生物碱(steroidal glycoalkaloids, SGAs)是茄科和百合科等许多植物主要的次生代谢产物。它具有强大的溶解特性和抑制乙酰胆碱酯酶活力[1-2]。它可以麻痹呼吸系统和运动系统中枢神经,并有溶血和刺激、腐蚀粘膜的作用,甚至有致畸和致死的可能。在马铃薯(Solanumtuberosum)中,当SGA含量超过200 mg/kg FW时,严重影响食味并且威胁食品安全性[3]。但是,SGAs能阻止动物或昆虫的取食及微生物的攻击[4-5],这为育种家培育抗病虫害的品种奠定了良好的基础。
糖苷生物碱的含量与外界条件有一定的关系。当温度达到10℃时,长期贮藏的马铃薯块茎中的SGAs含量可以增加到518 mg/kg FW[6],而且随着贮藏时间的延长,特别是贮藏期间见光,薯皮变绿,SGAs的含量明显增加。本课题组在前期的研究中发现红光对块茎SGAs含量有明显的诱导作用,其次为蓝光[7]。在马铃薯栽培种中α-茄碱和α-查茄碱是2个天然存在的主要类固醇糖苷生物碱,占总生物碱苷类的95%,这两种生物碱具有协同增效作用,被糖苷生物碱合成代谢支路的末端茄啶糖基转移酶 (solanidine glycosyltransferases) 催化。目前,已鉴定并克隆出的3个糖基转移酶[8],分别是茄啶半乳糖基转移酶 (solanidine galactosyltransferase, SGT1)、茄啶葡萄糖基转移酶 (solanidine glucosyltransferase, SGT2)和鼠李糖转移酶 (rhamnosyl transferase, SGT3),其中SGT1和SGT2分别催化产生γ-茄碱和γ-查茄碱,SGT3催化β-茄碱和β-查茄碱转化为其相应的α形式[9-10]。为了进一步研究此类基因的表达特点以及调控机制,Rockhold等[11]已经克隆到SGT2启动子,并将其构建β-葡糖醛酸酶(β-glucuronidase, GUS)融合表达载体导入到马铃薯中,发现叶片中的GUS的表达量明显高于块茎中的,但均低于阳性对照CMV 35S (cauliflower mosaic virus) 启动子驱动的转基因植物。对于马铃薯SGT1和SGT3两种糖基转移酶的调控与启动子功能的研究尚未见报道。
本研究拟通过红光诱导处理,研究马铃薯糖苷生物碱合成代谢关键酶SGT3的基因光诱导表达特点;确定并克隆SGT3基因启动子序列,通过用PLANTCARE和PLACRE软件对启动子序列进行分析,构建不同缺失片段启动子与报告基因GUS的融合基因,进行烟草(Nicotianatabacum)的瞬时表达和稳定遗传转化,检测不同缺失片段启动子驱动的转基因植株中报告基因的表达水平,以此分析马铃薯SGT3基因启动子的功能。
1 材料与方法
1.1 植物材料
马铃薯栽培种庒薯3号的无菌组培苗、微型薯以及四倍体烟草品系T12均由甘肃省作物遗传改良与种植创新重点实验室提供。
1.2 菌株和质粒
大肠杆菌(Escherichiacoli) DH5α、农杆菌 (Agrobacteriumtumefaciens) LBA4404、表达载体pBI121(图1)均由本实验室保存。SGT3启动子(NCBI注册号:KC331037)。中间载体 pGEM®-T Easy 载体、限制性内切酶和T4 DNA 连接酶购自Promega公司。引物由生工生物工程(上海)股份有限公司(Sangon Biotech Co.,Ltd. Shanghai)合成。DNA凝胶回收试剂盒购自天根生化科技(北京)有限公司 (Tiangen Biotech Co., Ltd. Beijing)。其他生化试剂均为国产分析纯。

图1 表达载体pBI121T-DNA结构示意图Fig.1 T-DNA structure diagram of expression vector pBI121 RB,LB:T-DNA 的右边界序列和左边界序列 The right border sequence and the left border sequence of T-DNA;NOS-pro,NOS-ter:NOS基因启动子和终止子 NOS promoter and terminator;nptII(Kan’):卡那霉素抗性基因 Kanamycin resistance gene;HindⅢ, BamHⅠ:HindⅢ和BamHⅠ限制性内切酶 HindⅢ and BamHⅠrestriction sites;CaMV 35S:花椰菜花叶病毒基因的启动子 Cauliflower mosaic virus gene promoter;GUS:β-葡糖醛酸酶基因 β-glucuronidase gene;ATG,TGA:GUS基因的转录起始密码子和终止密码子 GUS gene transcription.
1.3 材料处理
用自来水冲洗试管微型薯,选择平均直径约为8 mm的微型薯分为2组,均保存在15℃黑暗培养箱。其中一组用锡箔纸包裹作为黑暗对照,另一组用15 W的红灯泡照射(19.6~19.7 mol/m2·s)。分别在6,12,24 h后收集样品并立即液氮处理后放在—80℃冰箱待用,以新鲜收获的微型薯作为对照。
1.4 总RNA提取及其反转录
从冷冻的微型薯中提取总RNA,具体操作根据 Simple Total RNA 试剂盒的说明 (Tiangen Biotech CO., LTD Beijing) 。总RNA 的浓度测定用 Ultrospec 1100 pro 分光光度计 (Amersham Biosciences),用260/280 nm 的吸光值检测RNA的质量,其完整性用1% 的琼脂糖胶电泳检测[12]。定量400 ng RNA 进行反转录,具体操作根据 PrimeScript®RT reagent Kit (TaKaRa Biotechnology Co., Ltd Dalian) ,其中包括基因组DNA的去除。
1.5 实时定量PCR
实时定量PCR以马铃薯延伸因子(ef1-α) 作为看家基因,马铃薯SGT3基因作为目的基因,其cDNA序列均从GenBank 数据库 (http://www.ncbi.nlm.nih.gov/)获得,引物设计用Primer Premier 5.0 (PREMIER Biosoft International) 软件完成(表1)。实时定量PCR用SYBR GreenⅠ作染料, Mx3005p Multiplex Quantitative PCR System (Stratagene) 完成整个实验,用20 μL反应体系,如下:10 μL 2×SYBR®Premix Ex TaqTM(TaKaRa Biotechnology Co., Ltd Dalian),0.4 μL ROX Reverence Dye (50X),1 μL cDNA,上下游引物的终浓度均为0.4 μmol/L,循环体系为:95℃ 30 s,40个循环(95℃ 5 s, 60℃ 30 s),以溶解曲线检查产物扩增的特异性:95℃ 15 s,60℃ 1 min,95℃ 15 s。所有样品重复3次,取平均值,实时定量PCR体系做2~3次重复以保证体系的稳定性。最终PCR产物用2% 琼脂糖胶电泳分离,证实产物的特异性。 CT 值通过Mx3005p Multiplex Quantitative PCR System 得出,用Excel 2003进行数据分析,SPSS 17.0 软件对数据进行邓肯法多重性显著性分析。相对表达量用ΔΔCT法计算[13]。即相对表达量=2-ΔΔCT,其中ΔΔCT=CT (未知—看家基因)—CT (对照—看家基因)。

表1 实验所需引物序列Table 1 Primer sequences used for this work
注:其中ef1-α和SGT3分别代表实时定量的内参基因引物和目的基因引物。P1,P2,P3,P4和P5分别代表5个缺失片段的上游引物和1个下游引物(R)。下划线代表酶切位点HindⅢ和BamHⅠ,前面为保护碱基。
Note: ef1-α and SGT3 represents the reference gene primer and the target gene primers respectively. P1, P2, P3, P4 and P5 stand for five missing fragment forward primer and a common reverse primer. Underlined sequences indicate the HindⅢ and BamHⅠ sites。
1.6 马铃薯SGT3基因启动子的序列分析
启动子序列分析采用PLANTCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) 和PLACRE (http://www.dna.affrc.go.jp/PLACE/signalup.html) 软件分析。
1.7 5′端各缺失片段的获得和T中间载体的构建
利用Primer Premier 5.0 (PREMIER Biosoft International) 软件设计5个缺失片段上游引物(P1,P2,P3,P4和P5)和1个通用下游引物(R)(表1),为方便后续表达载体构建,在上游引物和下游引物5′分别插入HindⅢ (CCCAAGCTT) 和BamHⅠ (CGGGATCC) 酶切位点(表1)。以SGT3启动子全序列质粒DNA为模板,按以下条件进行PCR扩增:反应体积为 20 μL,包括 1 μL DNA 模板,10 μL 10×DNA Mix,1 μL上游引物(10 mmol/L),1 μL 下游引物(10 mmol/L),7 μL ddH2O;反应条件为:95℃预变性 5 min;95℃ 5 s,55℃ 1 min;72℃ 1 min;35个循环后于72℃保温延伸10 min,得到5个目标片段 (349,572,979,1312 和1870 bp)。经1%琼脂糖凝胶电泳分离后用DNA凝胶回收试剂盒回收纯化,将得到不同长度的PCR产物连接到pGEM®-T Easy载体,经过蓝白斑筛选,通过PCR检测和酶切检测 (HindⅢ和BamHⅠ) 选择合适的菌体,测序结果用DNAMAN软件进行序列比对,其相似性均达到99%以上。将测序合适的5个菌体(P349,P572,P979,P1312和P1870)提取质粒DNA,用HindⅢ/BamHⅠ双酶切,得到5个小片段;同时用这两种酶双酶切pBI121质粒,得到切去 CMV 35S 启动子的 pBI121 大片段。将5个小片段分别和pBI121大片段连接,得到不同长度的SGT3p/GUS的5个植物表达载体,分别命名为P349, P572,P979,P1312和 P1870 (图2)。以CMV 35S驱动的pBI121作为阳性对照。该载体导入感受态大肠杆菌DH5α细胞,可利用pBI121上的nptII基因的卡那霉素抗性进行重组体转化菌的筛选。用HindⅢ和Bam HI双酶切验证该重组体。
1.8 烟草的稳定遗传转化和瞬时表达
采用农杆菌介导的烟草叶盘法[14]将构建好的5个缺失片段表达载体(P349, P572,P979,P1312和P1870)进行遗传转化,通过生根筛选,1个月后取顶部第 3 个叶片进行GUS组织化学染色,以非转基因烟草为阴性对照 (CK-),CMV 35S启动子驱动的GUS基因的pBI121转化烟草作为阳性对照 (CK+)。瞬时表达将共培养完毕后的烟草叶片在加入250 mg/L的头孢无菌水中脱菌2 h,进行GUS组织化学染色,照相并观察。以非转基因烟草叶片为阴性对照(CK-),CMV 35S启动子驱动的GUS基因的pBI121转化烟草作为阳性对照 (CK+)。
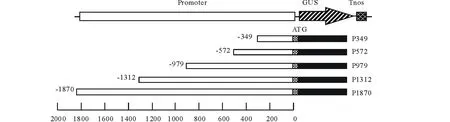
图2 SGT3基因启动子驱动报告基因GUS的融合表达载体构建Fig.2 Construction of GUS fusion expression vector driven by SGT3 promoter
1.9 GUS组织化学染色
参照张宁[15]的方法进行GUS染色,将转基因烟草的顶部第 3 个叶片、茎和根进行GUS染色,至少选取5株独立植株进行鉴定。37℃保温过夜,用70%的乙醇冲洗4~5次,待阴性对照为白色,将植物的茎切取横截面,放在载玻片上在正置万能显微镜(OLYMPUS BX61)下进行观察并照相,植物的叶片和根可以直接放在载玻片上观察。以非转基因烟草为阴性对照(CK-)。本实验于2012年4月-2013年3月在甘肃农业大学作物遗传改良与种质创新重点实验室完成。
2 结果与分析
2.1 红光显著诱导SGT3基因的表达
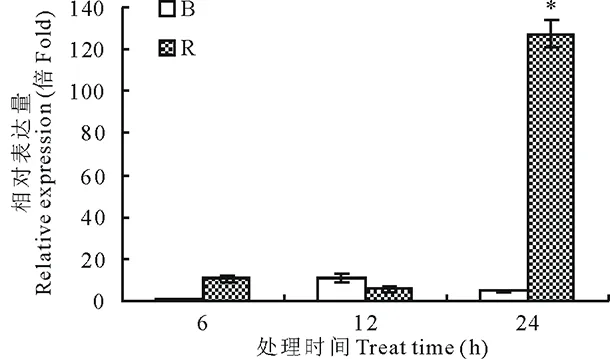
图3 SGT3基因表达特点Fig.3 The SGT3 gene expression B表示暗培养;R表示红光诱导;*表示在0.05水平差异显著。B denotes the dark treatment; R denotes red light induced; * indicates a significant difference at the 0.05 level.
将收获的微型薯通过红光诱导,用实时定量PCR方法测定在处理后6,12和24 h时SGT3基因的相对表达量。结果显示,SGT3基因在15℃黑暗处理12 h,其微型薯的mRNA水平有所升高,但随着时间的增加并无上升趋势,在处理24 h后反而有所降低,说明在黑暗处理下,随着时间的变化SGT3的含量没有明显改变。在红光诱导6 h后,与黑暗对照相比,SGT3的表达量增加,T-test结果表明在0.05水平上其差异不显著;在红光诱导24 h后,SGT3的转录水平较黑暗对照增加了26.8倍,且在0.05水平上差异显著,这说明红光明显诱导SGT3基因的表达,尤其在红光诱导24 h后表达量显著增加(图3)。
2.2 SGT3启动子的序列分析

图4 扩增SGT3基因启动子Fig.4 Amplification result of SGT3 gene promoter

图5 SGT3基因的启动子序列Fig.5 Sequence of SGT3 gene promoter 下划线表示构建缺失片段的5个上游引物和1个下游引物;ATG的A记为+1位点,起始密码子ATG,TATA-Box,CAAT-Box均用灰色表示,部分光调控元件用粗体表示。 →代表预测的转录起始位点。The underlined sequences are five forward primers and one reverse primer; the start codon,TATA-Box and CAAT-Box are highlighted in grey; some elements of light regulation are shown in bold; transcription start site is shown in black arrow →.
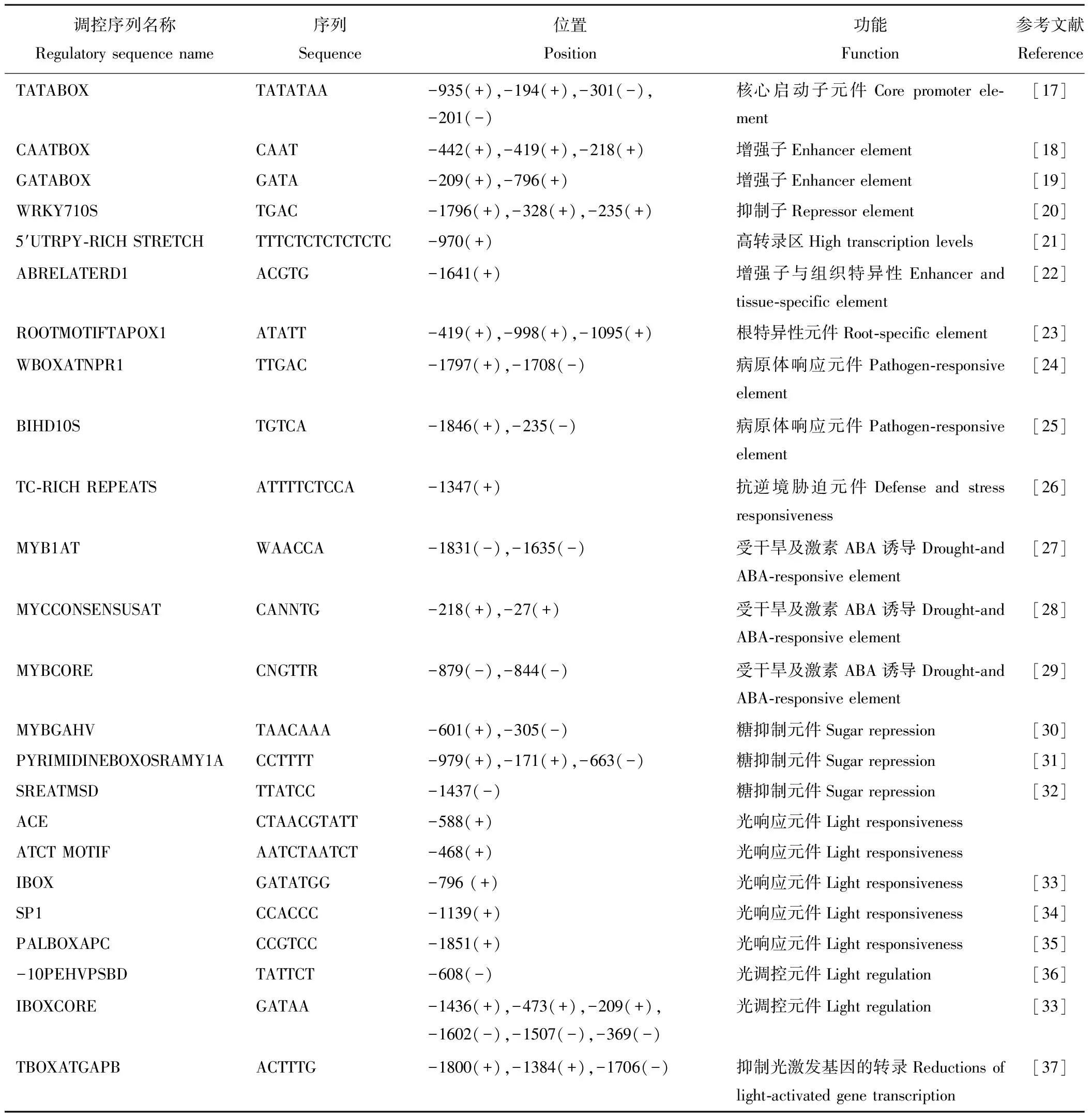
调控序列名称Regulatory sequence name序列Sequence位置Position功能Function参考文献ReferenceTATABOXTATATAA-935(+),-194(+),-301(-),-201(-)核心启动子元件Core promoter ele-ment[17]CAATBOXCAAT-442(+),-419(+),-218(+)增强子Enhancer element[18]GATABOXGATA-209(+),-796(+)增强子Enhancer element[19]WRKY710STGAC-1796(+),-328(+),-235(+)抑制子Repressor element[20]5′UTRPY-RICH STRETCHTTTCTCTCTCTCTC-970(+)高转录区High transcription levels[21]ABRELATERD1ACGTG-1641(+)增强子与组织特异性Enhancer and tissue-specific element[22]ROOTMOTIFTAPOX1ATATT-419(+),-998(+),-1095(+)根特异性元件Root-specific element[23]WBOXATNPR1TTGAC-1797(+),-1708(-)病原体响应元件Pathogen-responsive element[24]BIHD10STGTCA-1846(+),-235(-)病原体响应元件Pathogen-responsive element[25]TC-RICH REPEATSATTTTCTCCA-1347(+)抗逆境胁迫元件Defense and stress responsiveness[26]MYB1ATWAACCA-1831(-),-1635(-)受干旱及激素ABA诱导Drought-and ABA-responsive element[27]MYCCONSENSUSATCANNTG-218(+),-27(+)受干旱及激素ABA诱导Drought-and ABA-responsive element[28]MYBCORECNGTTR-879(-),-844(-)受干旱及激素ABA诱导Drought-and ABA-responsive element[29]MYBGAHVTAACAAA-601(+),-305(-)糖抑制元件Sugar repression[30]PYRIMIDINEBOXOSRAMY1ACCTTTT-979(+),-171(+),-663(-)糖抑制元件Sugar repression[31]SREATMSDTTATCC-1437(-)糖抑制元件Sugar repression[32]ACECTAACGTATT-588(+)光响应元件Light responsivenessATCT MOTIFAATCTAATCT-468(+)光响应元件Light responsivenessIBOXGATATGG-796 (+)光响应元件Light responsiveness[33]SP1CCACCC-1139(+)光响应元件Light responsiveness[34]PALBOXAPCCCGTCC-1851(+)光响应元件Light responsiveness[35]-10PEHVPSBDTATTCT-608(-)光调控元件Light regulation[36]IBOXCOREGATAA-1436(+),-473(+),-209(+),-1602(-),-1507(-),-369(-)光调控元件Light regulation[33]TBOXATGAPBACTTTG-1800(+),-1384(+),-1706(-)抑制光激发基因的转录Reductions of light-activated gene transcription[37]
为了探索SGT3基因的光诱导原理,本实验将克隆到2449 bp的潜在SGT3启动子(图4箭头所示),通过序列分析发现该片段的3′端与已报道的SGT3基因cDNA 5′端完全相同(57 bp),将此序列在GenBank数据库中进行Blast对比,发现此启动子序列为首次克隆到,并且与马铃薯染色体11上的RH170D10同源性达到98%,说明克隆到的是SGT3 编码起始密码子ATG的上游序列。通过Core promoter软件(http://rulai.cshl.org/tools/genefinder/CPROMOTER/human.htm)在线预测该启动子的起始位点,初步确定转录起始位点为翻译起始密码子上游-152 bp处的 “A” 碱基。将SGT3基因启动子序列在植物顺式作用元件数据库 PLANTCARE和PLACE中进行生物信息学分析,发现在翻译起始密码子上游-194和-935处可能存在2个TATA-box,同时确定了3个CAAT-box的位置(图5),在该启动子序列-970处有1个赋予SGT3基因高转录水平(high transcription levels)的5′UTR Py-rich stretch元件和3个糖抑制元件(Sugar repression)(表2)。研究表明SGT3基因的合成受到多种环境因素调控[16],本实验通过数据库搜索,预测到SGT3基因启动子中含有多个受干旱和受ABA诱导 (drought-和ABA-responsive)、病菌诱导(pathogen-responsive) 等调控元件,同时对启动子的组织特异性元件(tissue-specific element)、增强子元件(enhancer element) GATAbox、抑制子元件(repressor element)也进行了预测, 另外在此DNA序列及其互补链中分布着许多与光调控相关的顺式作用元件, 其中包括典型的光应答元件:ACE,ATCTMOTIF,IBOX,-10PEHVPSBD等 (表2),但对于这些调控元件的功能还有待于进一步的实验证明。
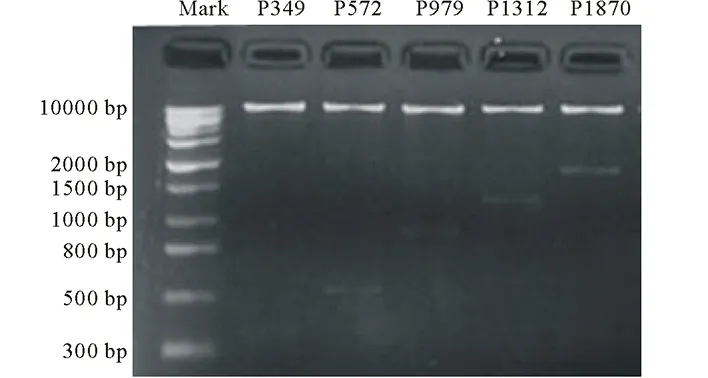
图6 不同缺失片段表达载体双酶切鉴定图Fig.6 Double digestion detection of expression vector driven by SGT3 promoter
2.3 不同长度的SGT3启动子驱动GUS基因在烟草愈伤组织中的瞬时与稳定表达
利用烟草叶片浸染法[38]分别将构建好的植物表达载体P349, P572,P979,P1312、P1870(图6)及CMV 35S启动子驱动的阳性对照导入烟草愈伤组织。通过GUS组织化学染色发现,5种不同长度的SGT3都能驱动GUS基因的表达,但表达水平存在差异,在P572,P979的表达强度较高,在 P1312和P1870明显减弱。而且不同长度SGT3启动子驱动的愈伤组织的GUS表达水平没有阳性对照CMV 35S驱动的表达量高,非转化愈伤组织中无GUS表达(图7A)。
为了进一步分析SGT3基因在烟草中的稳定遗传表达,采用叶盘法[14]将5个不同缺失片段的GUS融合表达载体导入烟草叶片,经过愈伤组织分化和生根筛选,得到稳定表达的烟草组培苗。选取顶部第 3 个叶片进行GUS染色。结果显示,所有转基因烟草叶片均变蓝,未转基因烟草叶片不变色,说明阳性对照及其5种不同长度SGT3驱动的GUS基因均成功转入烟草,但其表达水平存在显著差异。其中P572片段驱动的转基因烟草GUS表达量较高,P1870片段驱动的转基因烟草表达量较低。但5种不同长度的SGT3驱动GUS转基因烟草的表达均没有阳性对照pBI121中CMV 35S启动子驱动的表达量高(图7B),这与瞬时表达结果一致。这证明了SGT3基因启动子和CMV 35S启动子已转化进入到烟草植株基因组中,并能在正常光照条件下诱导驱动GUS基因的表达,说明SGT3启动子的活性没有CMV 35S启动子的活性强。
2.4 不同组织中SGT3驱动GUS基因的特异性表达
通过烟草的瞬时表达和遗传稳定表达,发现P572片段驱动的GUS在转基因烟草中的表达量较高,将其顶部第 3 个叶片、茎和根进行GUS染色,放在正置万能显微镜下观察其组织特异性。在烟草叶片中,GUS染色主要集中在烟草叶片的叶脉部分,而叶肉细胞GUS染色不明显(图8C)。茎中GUS染色主要分布在表皮、韧皮部及木质部,其中GUS表达量在木质部最高,在髓部几乎不表达(图8B)。根中主要分布在根冠、分生区以及维管束组织中(图8A)。以上结果说明SGT3基因启动子驱动的GUS基因主要表达在分生组织或维管束组织中。
3 讨论
SGT3作为糖苷生物碱末端合成酶,对调控茄碱的表达有至关重要的作用,而启动子是精确调控外源基因在植物体内表达的重要元件[39]。本实验首先通过红光诱导,发现当红光和黑暗同时处理马铃薯试管薯6~12 h后,SGT3的表达量没有显著变化。 而当红光处理24 h后,SGT3的表达量显著增加,而且研究发现红光显著诱导SGA含量的增加[7],因此红光可能诱导了SGT3基因的表达进而导致SGA含量增加。
本项研究的结果同时暗示了糖苷生物碱在光下含量高[7]的原因可能与SGAs合成代谢相关基因的调控密切相关。为进一步探究其调控原理,首次从马铃薯栽培种克隆到SGT3启动子,通过信息学分析初步预测该启动子的起始位点位于翻译起始位点上游-152 bp处,其准确性则需要通过进一步试验验证。序列分析发现SGT3启动子序列存在大量光调控元件,如ACE,ATCTMOTIF,IBOX,-10PEHVPSBD。烟草的愈伤组织瞬时表达和烟草稳定遗传结果表明:不同长度SGT3驱动的GUS基因均在烟草的愈伤组织和转基因叶片中表达,但其表达强度没有阳性对照pBI121的表达量高,说明所研究SGT3启动子的驱动活性没有启动子CMV 35S的活性强,研究表明马铃薯SGT2启动子驱动的转基因植株的表达量也没有CMV 35S启动子驱动的转基因植株的表达量高[11]。在缺失片段驱动的GUS烟草愈伤组织和转基因叶片中发现,P572和P979表达量较高, 序列分析结果表明在该片段存在加强子(-796)和高转录区(-970),以及光调控元件ACE(-588)和IBOX(-796)。已知ACE和IBOX是光诱导基因启动子中普遍存在的光应答元件(LREs),它们对光调控的转录激活是必需的[40-42],这可能是P572和P979驱动的GUS基因表达量高的原因,也说明此段区域是启动子调控的关键序列部分,预测结果与瞬时表达和烟草的稳定表达结果均一致。而P1312和P1870的表达量减弱,序列分析发现,在此片段存在抑制子(-1796)和 TBOXATGAPB(-1800和-1384),据报道TBOXATGAPB是抑制光激活基因转录的元件[37],它能有效地调控光调控元件的表达,同时由于启动子自身序列长度的增加,减弱了启动子的活性。SGT3基因的启动子与GUS组成的融合表达基因(SGT3p/GUS)在烟草的分化部位以及疏导组织中有比较高的表达,可能与这些部位特殊的疏导作用相关,其疏导作用最终亦要以跨膜运输的方式进行,而通过TMHMM 2.0软件对SGT3蛋白的跨膜区预测发现,该蛋白在50AA处存在跨膜结构。这进一步证实了SGT3启动子表达的组织特异性。

图7 烟草叶片不同缺失片段GUS染色Fig.7 GUS expression in tobacco driven by the promoters of SGT3 and CMV 35S A.瞬时表达;B.T1代。CK- 代表未转基因烟草叶片;CK+、P349、P572、P979、P1312和P1870分别表示转pBI121、P349、P572、P979、P1312和 P1870的烟草叶片GUS 组织化学检测结果。A: Transient expression; B: T1 generation. CK- represents not genetically modified; CK+, P349, P572, P979, P1312 and P1870 shown GUS histochemical assay of tobacco leaf transformed with P349, P572, P979, P1312 and P1870, respectively.
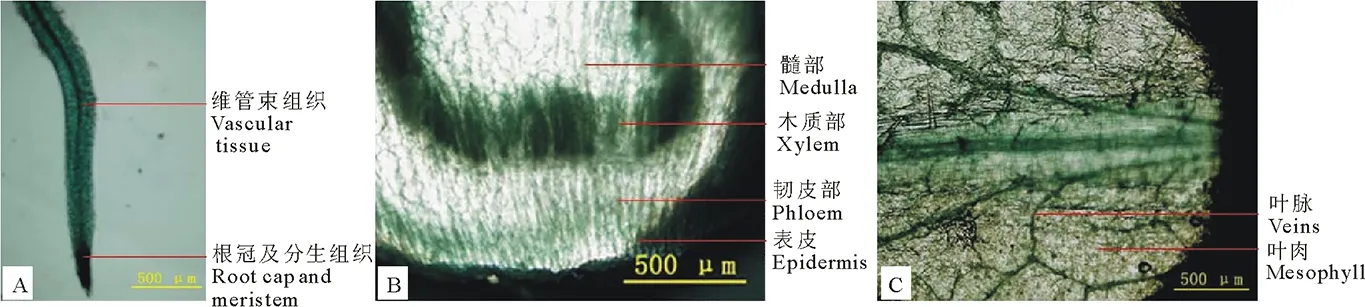
图8 P572驱动的GUS基因在烟草根、茎和叶片中的稳定表达Fig.8 Stable expression of GUS gene in root、stem and leaves driven by P572A.根 Root; B.茎 Stem;C.叶片 Leaf.
瞬时表达不能将转染的核酸整合到染色体上,结果只是短暂的高水平的表达,因此也就无法精确定位转染核酸表达的位置,同时由于DNA的摄入效率和表达水平在不同试验中差异较大,不长久也不稳定,得到的蛋白质保存时间也比较短。本实验通过比较烟草叶片侵染瞬时表达和稳定遗传表达,发现瞬时表达还是有一定的可靠性,在烟草叶片中不同长度SGT3启动子驱动的GUS基因的瞬时表达和叶盘转化法的烟草稳定表达结果基本一致,但是烟草稳定表达更能确定地从植物遗传学角度去分析问题,它将核酸和染色体整合到一起,但整合并不一定意味着表达,只有整合到表达区的基因才会表达,而且整合到不同的染色体区段的外源基因表达的量也是不同的,所以还需通过生根筛选等新表型筛选稳定转染体,得到稳定表达的植株。
目前,已得到以上5种不同长度SGT3基因上游序列驱动的GUS基因表达的转基因植株,之后可以对各转基因植物进行光诱导和干旱、ABA、真菌侵染以及糖抑制等启动子活性检测,以便对该启动子功能做更细致的分析,从而进行启动子的合理改造, 如在保留其核心元件和功能元件的前提下,改变其组织特异性功能元件,提高其表达活性,更好地应用于生物工程研究中,获得叶片中SGAs含量高而块茎中其含量低的优良马铃薯品种。
猜你喜欢
学与玩(2022年10期)2022-11-23
今日农业(2022年3期)2022-06-05
浙江中西医结合杂志(2017年2期)2017-01-12
现代工业经济和信息化(2016年2期)2016-05-17
当代化工研究(2016年9期)2016-03-20
创新科技(2015年1期)2015-12-24
电子工业专用设备(2015年4期)2015-05-26
汽车维修与保养(2015年8期)2015-04-17
无机化学学报(2014年3期)2014-02-28
声屏世界(2014年6期)2014-02-28
