
基于氢氘交换核磁共振技术研究蛋白质与阳离子交换介质的相互作用
2014-10-31 10:31王康郝冬霞齐树亭马光辉
生物工程学报 2014年9期
王康,郝冬霞,齐树亭,马光辉
1 河北工业大学海洋科学与工程学院,天津 300130
2 中国科学院过程工程研究所 生化工程国家重点实验室,北京 100190
随基因工程等现代生物技术的迅猛发展,以细胞为工厂生产疫苗、抗体、酶、细胞因子等蛋白产品已成为趋势。细胞产物的复杂性使高效柱层析成为蛋白产品分离纯化的支撑技术,其中,离子交换层析又是蛋白质分离纯化中最常用的手段之一[1-2]。然而,由于蛋白分子易失活,常造成层析中洗脱峰的展宽、拖尾及肩峰,最终造成蛋白回收率、选择性和柱效显著下降等诸多问题,因此,蛋白活性保持困难仍然是目前离子交换层析中普遍面临的瓶颈问题。
蛋白质的活性保持和选择性机制取决于蛋白与层析介质的相互作用过程。因此,探索蛋白在层析介质上的结构变化是一直以来的研究热点。传统研究多从蛋白的宏观保留行为 (保留时间、收率、活性)上来推测不同介质及操作条件对蛋白与介质微观吸附过程的影响[1,3-4]。近年来,随着新型微观界面分析仪器的出现及组合联用,该领域有望上升至分子层次上研究蛋白质在层析介质表面的结构变化[5-8]。在目前已建立的蛋白质在液固界面的微观表征手段中,如荧光光谱 (FR)、圆二色谱 (CD)、原子力 (AFM)、双偏振极化干涉仪 (DPI)、质谱 (MS)等[9-13],已可获取大量蛋白分子水平信息包括蛋白质吸附厚度、形貌特征、聚集状态、吸附取向等。但是由于这些技术大多局限于芯片平面材料或纳米球材料上检测,原位实时捕捉多孔介质内的蛋白分子结构变化信息至今仍很难实现。
据此,本课题组设计了一条蛋白质氢氘交换反应与核磁共振技术相结合的新型蛋白质液固界面表征路线,并在前期成功地表征了不同配基密度的疏水介质诱导的蛋白质去折叠行为[14]。本研究利用这种新型蛋白质液固界面表征手段。以离子交换色谱过程为研究对象,阳离子交换介质 (SP Sepharose FF)为模型介质,溶菌酶为模型蛋白。探索酰胺的氢氘交换对于表征蛋白质结构变化的可行性,并获取溶菌酶在介质表面吸附时的去折叠行为以及绑定位点变化。以期为进一步理解蛋白质与离子交换介质微观作用机制,以及蛋白质分离纯化条件及层析介质结构的精确设计和定向优化提供理论依据。
1 材料与方法
1.1 材料与试剂
蛋清溶菌酶 (Hen egg white Lysozyme)购自 Sigma公司;阳离子交换凝胶介质 (SP Sepharose FF)购自GE Healthcare公司;其他所用试剂均为分析纯。
1.2 核磁共振波谱仪及参数
600 M核磁共振波谱仪 (Bruker Ascend-600 Avance Ⅲ),TCI三共振反式超低温探头。一维核磁共振氢谱 (1H-NMR)采用的脉冲程序为noesygppr1d,采样 16次;二维氢氢全相关谱(2D-1H-1H TOCSY)采用的脉冲程序为dipsi2gpph19,采样24次;采样前进行90°脉冲校准以及水峰的压制。
1.3 吸附蛋白的氢氘交换标记
蛋白在阳离子交换介质上的吸附以及标记路线如图 1所示。首先,阳离子交换介质 SP Sepharose FF装填成柱体积为1 mL的色谱柱,用起始缓冲液 (20 mmol/L PBS, pH 7.0)平衡色谱柱;然后,蛋白质溶解到起始缓冲液配成0.21 mmol/L溶液,进样5 mL,缓冲液淋洗并且检测紫外吸收值,当紫外吸收值为0时,停止淋洗。其次,用标记缓冲液 (D2O,20 mmol/L PBS,pD 7.0)流经色谱柱,15 min后通入淬灭缓冲液(D2O,20 mmol/L NaAC,pD 3.0)终止氢氘交换反应。最后,已经标记完的蛋白用洗脱液 (D2O,20 mmol/L NaAC,1 mol/L NaCl,pD 3.0)洗脱,所获的标记蛋白用 3 kDa的超滤膜脱盐冻干之后核磁共振检测。对照组样品 (没有吸附的蛋白)的做法和上述步骤相似,除去不添加吸附阳离子交换介质以及洗脱液。
1.4 氢氘交换数据分析
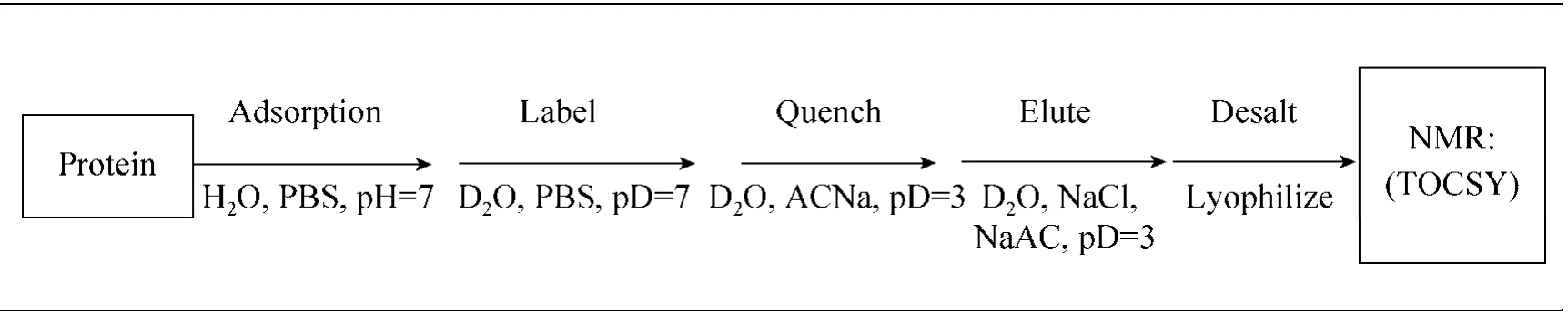
图1 蛋白质在阳离子交换介质上吸附、标记及检测路线Fig. 1 The adsorption, labeling and NMR analysis of protein on cation exchange media (SP Sepharose FF).
溶菌酶氨基酸残基核磁共振信号根据与 α位碳相连的酰胺氢 (NH-CαH)来归属,并以文献报道[15]结合生物大分子核磁共振数据库Biological Magnetic Resonance Bank (BMRB 4562)为参考。峰强度 (I)通过软件 Topspin version 3.0标定。为了排除样品浓度的影响,以不发生氢氘交换的芳香族氨基酸Trp108的 H4-H5相关峰为参考峰,对特征残基峰的强度进行归一化处理,如式 (1):

式中Ipeak为残基峰的强度;Ireference为参考峰的强度;Inomalize为归一化后的峰强度。通过比较不同状态归一化后蛋白残基峰强度的变化可以获取蛋白各个残基酰胺氢对溶剂可及的敏感程度以及酰胺氢被保护程度等信息[16-17]。
1.5 溶剂可及表面计算
通过分子模拟软件Accelrys discovery studio(Accelrys, Inc. San Diego, USA)计算蛋白各个残基的溶剂可及表面百分比 (Solvent accessible surface area,SASA)。溶菌酶的三维结构来源于蛋白质结构数据库 (PDB code: 1E8L),探针半径为 1.4 Å。
1.6 蛋白静电势计算
蛋白质静电势采用通用算法程序,利用分子模拟Accelrys discovery studio软件中Delphi模块进行蛋白静电势的模拟计算[18-19]。
2 结果与分析
2.1 溶菌酶的氢氘交换特征
蛋白质氢氘交换技术是探测溶液中蛋白质的结构稳定性、折叠动力学和相互作用的重要研究工具,但在蛋白质液固界面吸附中应用较少。为确定溶菌酶在液固界面研究中的交换条件和可行性,首先考察了溶菌酶的氢氘交换特征,通过测定溶菌酶在不同状态下1D1H-NMR谱及其酰胺氢峰强度随时间的变化,来评估蛋白结构对氢氘交换的敏感性以及交换反应的合理时间。
结果如图 2所示,测定了溶菌酶在天然态(90% H2O/10% D2O,pH 7.0,25 ℃)和变性态(60% ACN/40% D2O,pH 7.0,25 ℃)不同时间间隔点的一维核磁共振氢谱 (1D1H-NMR)。其中,由于1D氢谱中大部分残基的谱峰叠加,单独选取了谱峰清晰分辨率较高的W123、W111、W63和 W108四个代表残基来观察溶菌酶在天然态和变性态的动力学变化。天然态 (图2,A-1、A-2)显示,4个残基的酰胺氢峰强度在开始10 min内快速下降,在20 min到180 min时则下降趋缓。对比变性态 (图 2 B-1、B-2),溶菌酶酰胺氢不但化学位移有显著变化,并且4个残基的峰强在更短时间约5 min内降到最低。这反映出天然态溶菌酶能够保持紧密三维球形状态,氘离子较难到达处于蛋白内部的残基区域,氢氘交换速率及峰强下降比较缓慢;而在变性剂乙腈溶液中,蛋白结构发生改变,致使个别残基峰周围的电子云环境改变而造成化学位移改变,并且由于蛋白去折叠,溶剂中的氘离子很容易到达去折叠的酰胺区域发生氘代反应,峰强下降且很快达到平衡。以上现象表明溶菌酶的结构变化对氢氘交换反应非常敏感,蛋白的去折叠程度可由其评估。此外氢氘交换在天然态和变性态下达到平衡的时间分别是10 min和5 min,因此在后续实验中蛋白在阳离子交换介质上吸附交换时间控制在10 min以上。

图2 不同溶液中,溶菌酶一维核磁共振氢谱 (A)及残基峰强度随时间的变化情况 (B)Fig. 2 1D IH NMR spectra (amide/aromatic region)of lysozyme in different solvents and the intensity change of selected residues (marked)as a function of time.
2.2 溶菌酶在阳离子交换介质表面的吸附行为
由于溶菌酶核磁信号在一维氢谱上严重重叠无法指认各个残基。因此本文拟通过二维氢谱中化学位移全相关谱TOCSY谱 (Total chemical shift correlation spectroscopy)来获得残基信息。TOCSY谱可以描述在蛋白肽链内具有α-H、β-H间偶极偶合相关的氢信号,其优点是以交叉峰的形式指认各个残基,谱峰很少重叠、分辨率较高。利用该技术,本文继续考察了溶菌酶在阳离子交换介质表面的吸附行为,根据各残基峰强度可以获知溶菌酶上氢质子被氘取代的程度,从而判断出蛋白质的去折叠程度。
在以上溶液实验的基础上,对比吸附态与天然态溶菌酶的TOCSY谱 (图3),显示吸附态时溶菌酶残基酰胺氢信号的丢失要多于天然态。说明吸附到阳离子交换介质上的溶菌酶暴露出更多的残基在重水溶剂里;同时,也说明溶菌酶紧密的球状蛋白开始松解,由此可推测阳离子交换介质表面的静电基团是介导溶菌酶去折叠行为的主要机制。
为详细了解溶菌酶蛋白各区域在阳离子交换介质表面的去折叠行为,对各个残基酰胺氢的交联峰进行归属和回归分析 (图4)。吸附态和天然态残基峰强度对比显示,吸附态残基峰强度明显比天然态残基弱,说明吸附态暴露出更多残基在溶液中。但是,绝大部分的残基信号保留下来,说明溶菌酶的结构只是发生局部去折叠,例如一些二级结构 α-helix、β-sheet (H1、H2、H3、H4、H5、H6,S2、S1除外)能够保持其原有结构。此外,不同的结构片段呈现出不同的氢信号保护程度,一些无规则卷曲 (Coil,bend,and turn)片段的氢信号就更容易失去 (如 69-75、80-95和115-120),相反,二级结构域对氢的信号保护则更好。以上研究表明,吸附态溶菌酶无规则片段去折叠程度更大,而二级结构域受到保护。
这种由静电作用介导的蛋白质去折叠与由疏水作用和变性剂介导的去折叠区域有许多相似之处[20-24],如二级结构被保护,无规则结构去折叠等。但去折叠程度明显不同,对比本课题组研究溶菌酶在苯基琼脂糖疏水介质表面的去折叠发现[14],静电配基介导的溶菌酶残基信号损失要明显少于后者,这说明静电作用导致的蛋白去折叠要弱于疏水作用对蛋白的破坏程度。
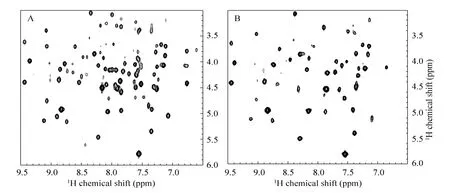
图3 溶菌酶的2D1H-1H-TOCSY谱 (A: 天然态;B: 吸附态)Fig. 3 2D 1H-1H-TOCSY NMR spectra of lysozyme. (A)Natural. (B)Adsorbed.
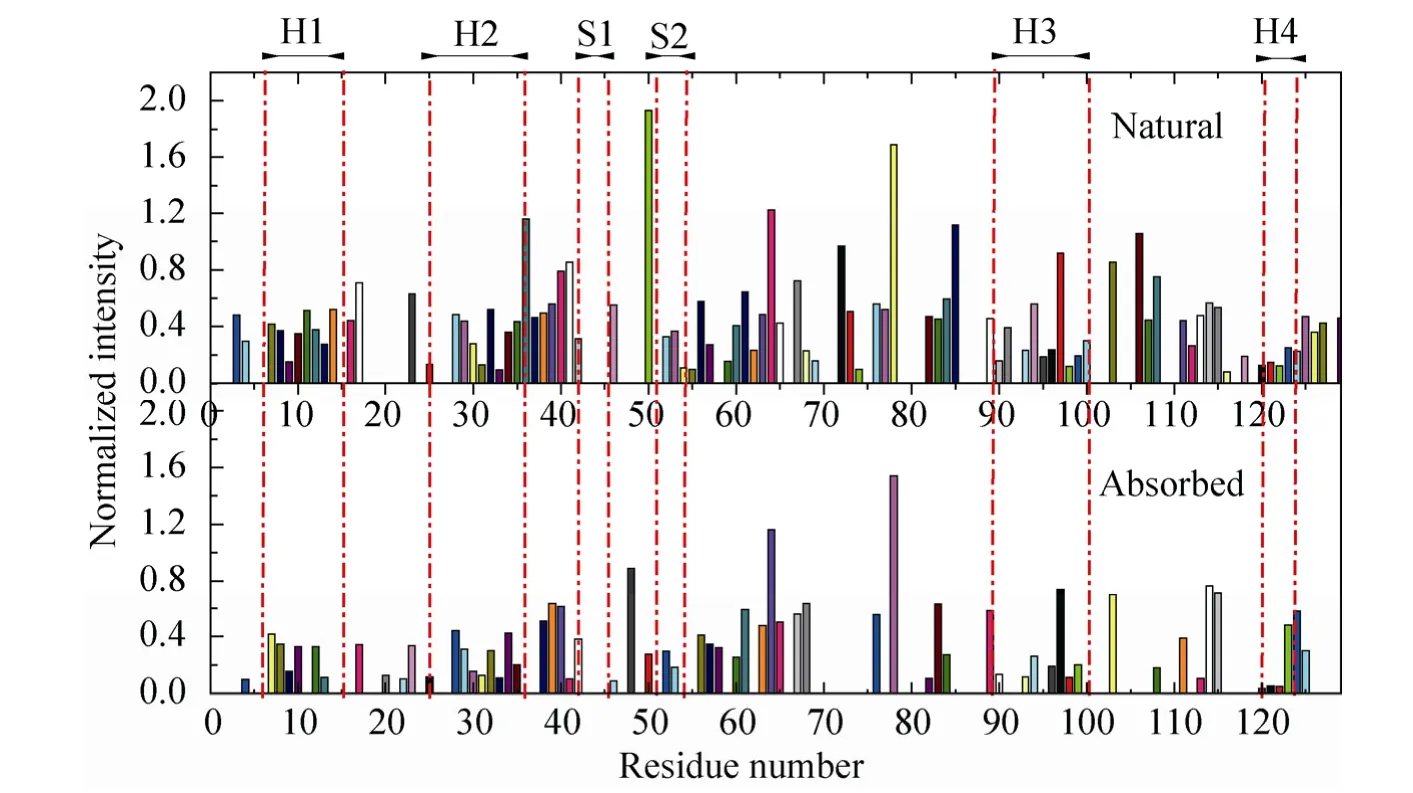
图4 天然态和吸附态溶菌酶氢氘交换行为的比较 (H=α螺旋,S=β折叠)Fig. 4 Comparison of the H/D exchange behavior between natural and adsorbed lysozyme. H=helix; S=sheet.
2.3 溶菌酶与阳离子交换介质的作用位点分析
详细了解蛋白质分子与介质相互作用中起决定作用的绑定位点,对于深刻理解层析过程中蛋白质与层析介质微观作用机理以及吸附剂的选择、设计具有重要意义。离子交换色谱利用蛋白质与离子交换介质之间的静电作用力的不同达到分离纯化。对于阳离子交换介质,由于带负电荷,主要由蛋白表面带正电荷的氨基酸与其发生静电作用。此外,研究者利用核磁共振检测到蛋白质与固体介质的作用界面的残基酰胺氢信号保护程度大于溶液中残基[25]。因此,可以确定蛋白与阳离子交换介质的作用位点主要有3个特征:1)带正电荷的氨基酸;2)蛋白表层的氨基酸;3)接触面酰胺氢信号被保护的氨基酸。本研究拟依据这3个特征确定溶菌酶与阳离子交换介质的结合位点。
首先,为确定蛋白表层带正电荷的氨基酸残基,通过计算机分子模拟计算软件 (Accelrys discovery studio)里的Delphi模块计算出每个残基的平均净电势,筛选出平均静电势大于0为带正电荷的氨基酸残基,然后,再从中筛选出SASA大于10%的残基,SASA大于10%的残基被认为是溶剂可及残基[26](图5)。
其次,通过公式 (2)筛选出被接触面保护的氨基酸。其中,I%为吸附前后蛋白的强度损失程度,当I%值大于0时即为酰胺氢信号被保护的残基 (图 6A)。图 6B为溶菌酶的氨基酸序列,其中被接触面保护的残基以斜体下划线标出,尽管这些氨基酸残基序列并不依次相连,但在蛋白三维结构中,大部分残基在同一表面。

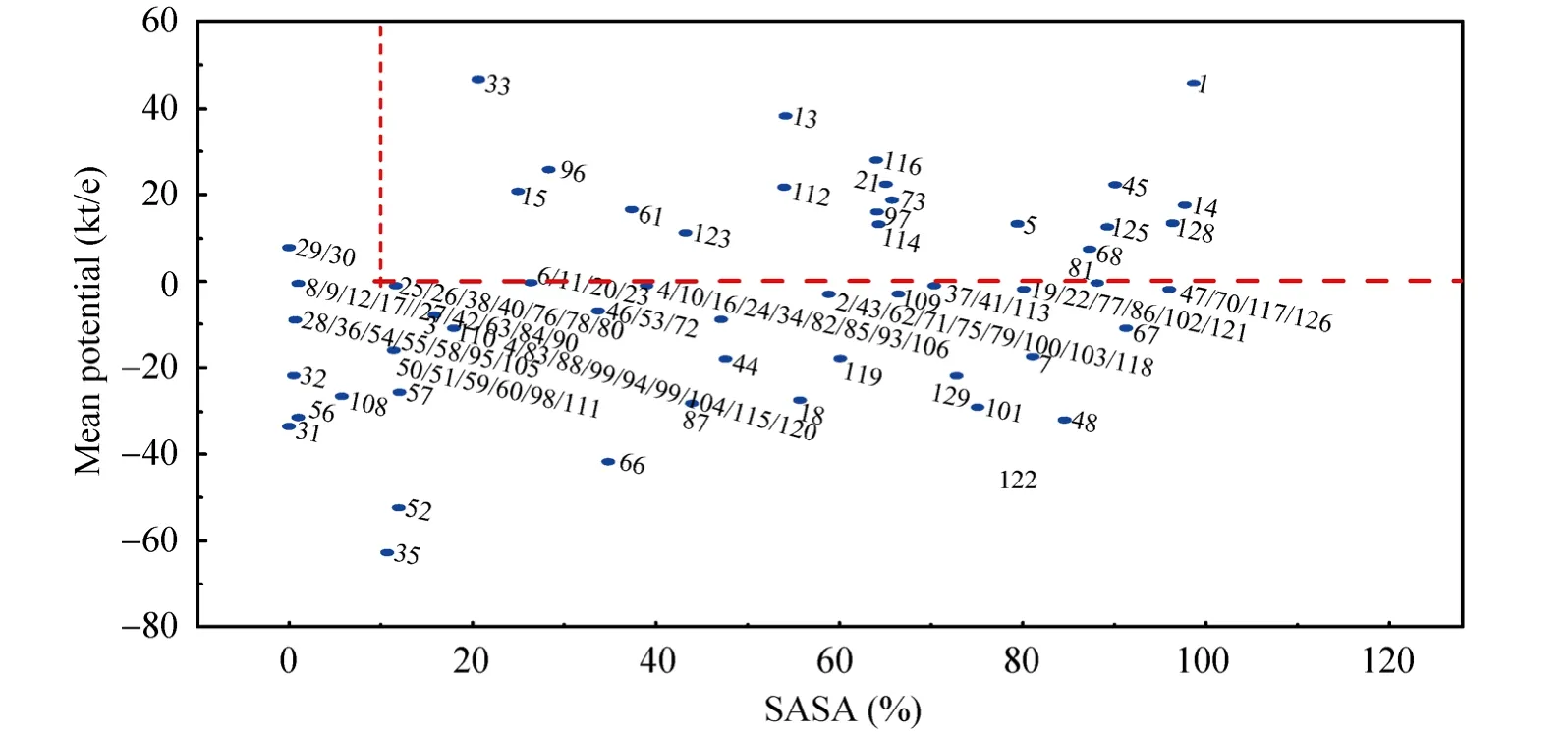
图5 平均电势和溶剂可及表面 (SASA)确定溶菌酶表面带正电荷的残基Fig. 5 Identif i cation of the exposed and positively charged residues on lysozyme according to residues’ mean potencial and SASA. The overlapping and approximate residues were labelled by‘/”.

图6 吸附态溶菌酶残基酰胺氢信号被保护程度 (A)和受到保护的氨基酸残基 (B)Fig. 6 Protection against H/D exchange of adsorbed lysozyme (A), I%>0=Protected residues, I%<0=Non-protected residues; Residues are protected against H/D exchange of adsorbed lysozyme (B), italic and underlined fonts.

图7 溶菌酶表面静电势分布及作用位点 (红色静电势小于零;蓝色静电势大于零)Fig. 7 Electrostatic potential distributions and possible sites of binding on the lysozyme, surface from–5.0 kt/e (red)to +5.0 kt/e (blue).
最终,取带正电荷的表层氨基酸与氢信号被保护的氨基酸之间的交集,筛选出溶菌酶与阳离子交换介质的作用位点分别为Lys33、Arg114和Arg68 (图 7),这与研究者用荧光标记赖氨酸测定的溶菌酶与相同阳离子交换介质 (即 SP Sepharose FF)的作用位点Lys33一致[27]。但由于荧光标记法只能标记赖氨酸一种带正电荷的残基,仅获得了赖氨酸的绑定信息。此外,另一类常用技术是通过比较定点突变蛋白的保留时间差异来判断介质表面的绑定位点[5,28-29],由于需要复杂的基因工程技术来构建蛋白质突变株,这些方法普遍存在如下问题:1)寻找存在突变株的蛋白较为困难,导致可研究的蛋白种类非常有限;2)没有考虑当引入两个或多个取代残基时蛋白三维结构改变对实验结果的影响。相比而言,本研究利用H/D交换结合核磁共振技术,能够弥补上述缺陷可获得所有残基包括带同样正电荷的精氨酸和组氨酸等的作用信息,并实现了原位标记、实时、快速的表征介质表面各种蛋白去折叠程度的信息。
3 结论
本研究利用氢氘交换结合 NMR技术表征了蛋白在离子交换介质上的吸附过程。结果显示,当溶菌酶吸附到介质表面之后,不同结构片段的去折叠程度不同,无规则卷曲 (Coil,bend,and turn)片段酰胺氢信号更易失去即去折叠明显,二级结构域 (α-helix,β-sheet)对酰胺氢信号保护较好即去折叠较弱,表明静电介导的蛋白去折叠呈局部分布。通过蛋白表面静电势模拟计算结合氢氘标记的蛋白核核磁数据分析确定了溶菌酶与阳离子交换介质的作用位点为Lys33、Arg114和Arg68。本研究提供了一条原位实时捕捉多孔介质内的蛋白结构变化信息的新途径,对于深刻理解层析过程中蛋白与层析介质微观作用机理具有重要意义,也为其他领域中蛋白质与生物材料之间微观界面研究提供了新的技术手段。
[1]DePhillips P, Lenhoff AM. Determinants of protein retention characteristics on cation-exchange adsorbents. J Chromatogr A, 2001, 933: 57–72.
[2]Yamamoto S, Nakanishi K, Matsuno R.Ion-exchange Chromatography of Proteins. New York: Marcel Dekker, 1988.
[3]Fogle J, Mohan N, Cheung E, et al. Effects of resin ligand density on yield and impurity clearance in preparative cation exchange chromatography. I.Mechanistic evaluation. J Chromatogr A, 2012,1125: 62–69.
[4]Gallant SR, Vunnum S, Cramer SM. Optimization of preparative ion-exchange chromatography of proteins: linear gradient separations. J Chromatogr A, 1996, 725: 295.
[5]Yao Y, Lenhoff AM. Electrostatic contributions to protein retention in ion-exchange chromatography.1. Cytochrome C variants. Anal Chem, 2004, 76:6743.
[6]Yao Y, Lenhoff AM. Electrostatic contributions to protein retention in ion-exchange chromatography.2. Proteins with various degrees of structural differences. Anal Chem, 2005, 77: 2157.
[7]Yoon BJ, Lenhoff AM. Computation of the electrostatic interaction energy between a protein and a charged surface. J Phys Chem, 1992, 96:3130.
[8]Zhou J, Chen SF, Jiang SY. Orientation of adsorbed antibodies on charged surfaces by computer simulation based on a united-residue model.Langmuir, 2003, 19: 3472.
[9]Wicar S, Mulkerrin MG, Bathory G, et al.Conformational changes in the reversed phase liquid chromatography of recombinant human growth hormone as a function of organic solvent:the molten globule state. Anal Chem, 1994, 66(22):3908–3915.
[10]Engel MFM, Visser AJWG, Mierio CPM van.Refolding of adsorbed bovine α-Lactalbumin during surfactant induced displacement from a hydrophobic interface. Langmuir, 2003, 19:2929–2937.
[11]Kim DT, Blanch HW, Radke CJ. Direct imaging of lysozyme adsorption onto mica by atomic force microscopy. Langmuir, 2002, 18: 5841–5850.
[12]Sonesson AW, Callisen TH, Brismar H, et al. A comparison between dual polarization interferometry (DPI)and surface plasmon resonance (SPR)for protein adsorption studies.Colloids Surf B Biointerfaces, 2007, 54(2):236–240.
[13]Xiao YZ, Jones TT, Fernandez EJ. Protein stability during HIC: hydrogen exchange labeling analysis and a framework for describing mobile and stationary phase effects. Biotechnol Bioeng, 2007,96: 80–93.
[14]Hao DX, Sandstrom C, Huang YD, et al.Residue-level elucidation of the ligand-induced protein binding on phenyl-argarose microspheres by NMR hydrogen/deuterium exchange technique.Soft Matter, 2012, 8: 6248–6255.
[15]Redfield C, Dobson CM. Sequential 1H NMR assignments and secondary structure of hen egg white lysozyme in solution. Biochemistry, 1988,27(1): 122–136.
[16]Bayraktar H, You CC, Rotello VM, et al. Facial control of nanoparticle binding to cytochrome C. J Am Chem Soc, 2007, 129: 2732.
[17]Keire DA, Gorenstein DG. Deriving structures from 2D NMR. A method for defining the conformation of a protein adsorbed to surfaces. Bull Magn Reson,1992, 14: 57.
[18]Oron A, Wolfson H, Gunasekaran K, et al. Using DelPhi to compute electrostatic potentials and assess their contribution to interactions. Curr Protoc Bioinfor, 2003, 8: 1–12.
[19]Haggerty L, Lenhoff AM. Relation of protein electrostatics computations to ion-exchange and electrophoretic behavior. J Phys Chem, 1991, 95(3):1472–1477.
[20]Bhattacharjya S, Balaram P. Hexafluoroacetone hydrate as a structure modifier in proteins:characterization of a molten globule state of hen egg-white lysozyme. Protein Sci, 1997, 6: 1065.
[21]Buck M, Radford SE, Dobson CM. A partially folded state of hen egg white lysozyme in trifluoroethanol: structural characterization and implications for protein folding. Biochemistry,1993, 32: 669.
[22]Jones TT, Fernandez EJ. α-Lactalbumin tertiary structure changes on hydrophobic interaction chromatography surfaces. J Colloid Interface Sci,2003, 259: 27–35.
[23]Sane SU, Cramer SM, Przybycien TM. Protein structure perturbations on chromatographic surfaces. J Chromatogr A, 1999, 849: 149–159.
[24]McNay JLM, Fernandez EJ. Protein unfolding during reversed-phase chromatography: I. effect of surface properties and duration of adsorption.Biotech Bioeng, 2001, 76: 224.
[25]Keire DA, Gorenstein DG. Deriving structures from 2D NMR. A method for defining the conformation of a protein adsorbed to surfaces. Bull Magn Reson,1992, 14: 57.
[26]Zloh M, Shaunak S, Balan S, et al. Identification and insertion of 3-carbon bridges in protein disulfide bonds: a computational approach. Nat Protoc, 2007, 2: 1070.
[27]Florian Dismer, Martin Petzold, Jürgen Hubbuch.Effects of ionic strength and mobile phase pH on the binding orientation of lysozyme on different ion-exchange adsorbents. J Chromatogr A, 2008,1194: 11–21.
[28]Gill DS, Roush DJ, Willson RC. Adsorption heterogeneity and thermodynamic driving forces in anion exchange equilibria of cytochrome b5. J Colloid Interf Sci, 1994, 167: 1.
[29]Gill DS, Roush DJ, Shick KA, et al.Microcalorimetric characterization of the anion-exchange adsorption of recombinant cytochrome b5and its surface-charge mutants. J Chromatogr A, 1995, 715: 81.
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
成都大学学报(自然科学版)(2021年1期)2021-05-22
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
世界农药(2019年3期)2019-09-10
世界农药(2019年3期)2019-09-10
池州学院学报(2015年3期)2016-01-05
天津科技大学学报(2015年2期)2015-08-09
火炸药学报(2014年3期)2014-03-20
中国海洋大学学报(自然科学版)(2014年6期)2014-02-28
中国兽药杂志(2012年4期)2012-11-06
