
手性硫脲催化剂不对称催化反应及氢键活化作用研究
2014-10-29 08:48陈盛楷赵志伟赵博文
西北民族大学学报(自然科学版) 2014年1期
陈盛楷,买 昊,赵志伟,赵博文,刘 颖,鲜 亮
(西北民族大学 化工学院,甘肃 兰州730124)
0 引言
不对称催化(包括化学催化和生物催化)是惟一具有手性放大作用的手性合成方法,在药物化学、材料科学以及合成化学等领域有着广泛的应用 .在不对称催化中,手性催化剂的设计合成是关键步骤.新型手性催化剂的出现,可以拓宽催化剂的底物适用范围,提高立体选择性,并用于合成手性药物或天然产物的手性砌块.而新型手性催化剂不对称催化反应机理或模型的研究,又用以指导设计合成更高效的手性催化剂.
自从Jacobsen等[1]首次发表了关于Strecker反应的手性硫脲的不对称催化反应以来,手性硫脲在不对称催化反应中的应用渐成热点(图1)[2].硫脲衍生物可以通过便宜易得的异硫氰酸酯偶联反应获得[3].原料易得、反应途径简单可靠、产率高等特点促进了手性硫脲在不对称催化领域中更加广泛的应用 .最近十年,有关该类反应的研究报道迅速增加,合成的手性硫脲催化剂的种类不断增多、结构更加复杂、涉及的反应类型也更加多样,对反应机理的研究也有了相当的进展 .这显示了有机不对称催化领域的研究人员对于该类手性有机小分子催化剂的研究兴趣日益增加[4~7].
1 手性硫脲不对称催化的氢键活化理论模型
氢键活化理论模型首先由Jacobsen等[8]提出,认为有机催化反应主要通过氢键共给体或非共价作用机理,利用氢键作用加速有机化学反应速率或者控制立体构型 .该类催化反应和Brφnsted酸催化反应并没有显著的区别 .手性硫脲及其衍生物的结构特点决定了氢键活化作用在不对称催化当中起到了关键作用(如图2所示).

图1 近年公开报道的一些典型手性硫脲催化剂

图2 硫脲衍生物的基本结构
硫脲结构中存在着N-H功能团,在反应中可以起到弱Brφnsted酸作用,能够和不同底物的亲核基团发生氢键相互作用.硫脲中硫羰基的硫原子HSAB软碱上的孤对电子可以和过渡金属Lewis酸发生配位作用[8],也能够和不同底物的亲电基团发生非共价键的相互作用 .硫脲化合物的这种酸碱双功能结构特点使得该类化合物无论对亲电试剂或亲核试剂都具有相当的活化作用[2].手性硫脲氢键活化的最大优势在于能够使得催化剂以多个催化位点和一个或两个底物同时发生非共价键的键合作用,从而活化底物 .在不对称催化反应中,硫脲的这些结构特点被利用来按照反应设计需求适当地组织催化反应作用位点,并控制催化反应选择性[4].
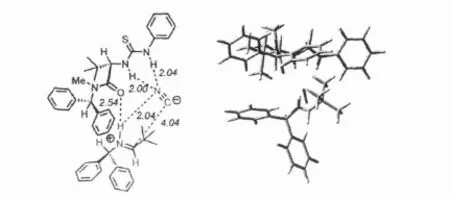
图3 手性硫脲双氢键催化亚胺氢氰化反应[9]
Jacobsen等[9]2009年研究了双氢键硫脲对映选择性催化亚胺氢氰化反应 .该反应中,氰根底物通过硫脲活泼氢形成了双氢键的中间体结构,而酰基氧也和亚胺底物形成了非共价氢键作用,从而促成了过渡态中间体的稳定性 .同时,手性硫脲催化剂的空间结构限制了过渡态中间体的空间结构,从而导致了对映选择性催化反应的发生.该氢键活化模型适用于绝大多数现在已知的手性硫脲对映选择性催化反应,如Mannich反应、Michael加成、aza-Henry反应、Strecker反应、Morita-Baylis-Hillman反应、Pictet-Spengler反应、aza-Diels-Alder反应和Petasis反应等[4].

图4 双氢键不对称催化模型的NMR和DFT研究[10]
2012年,王等[10]利用核磁共振及DFT计算等方法,展示了双官能团金鸡纳碱硫脲是如何在Michael反应中利用氢键作用对底物进行活化的 .在这其中,N-H分别和两个底物发生氢键作用,显著强化了催化剂-底物复合物的稳定性.DFT计算获得的对映选择性理论值(100%ee)和非对映立体选择性(60∶1dr)与实验测定值(98%ee和>30∶1dr)非常吻合 .这就证明了硫脲双氢键作用活化底物理论模型能够适用于该类反应.
在不对称催化反应研究中,绝大多数手性硫脲催化剂均在N上保留了两个活泼氢 .按照氢键活化理论,这种结构是有利于硫脲以Lewis酸性对底物产生选择性的氢键键合活化作用 .其中,应用最多的手性硫脲催化剂多具有刚性的苯基结构,并且在硫羰基相连的N上构筑了3,5-二(三氟甲基)苯基结构[11],而在另一侧的N上连接一个中心手性结构的取代基 .三氟甲基的存在能够通过调节硫脲主体结构的电子效应而影响催化剂对反应底物的非共价键合活化作用 .尽管Akiyama等[12]认为单氢键结构也能够对底物起到活化作用,但是,仅含有一个活泼氢的手性硫脲催化剂的不对称催化研究仍非常少见.

图5 含3,5-二(三氟甲基)苯基取代基团的手性硫脲催化剂[11]
在已报道的反应中,N原子上的活泼氢能够和F、Cl、Br和O等氢键的受体原子发生氢键键合作用,形成方向一定的分子间氢键结构 .这种结构对底物的结构和构象具有了一定的选择性,能够“迫使”底物以一定的方向和手性硫脲键合并被活化 .在此过程中,手性硫脲骨架结构另一侧的手性结构对底物也同样施加影响,从而促使底物以一定的构象与另外一个底物发生反应 .在这个过程中,含手性结构的取代基主要有两种方式对底物或者中间体和过渡态施加影响:其一是适当的空间位阻作用;其二,若取代基上存在着氢键的供体或受体原子,能够施加非共价键键合作用,从而对产物的立体结构产生积极的作用[13].
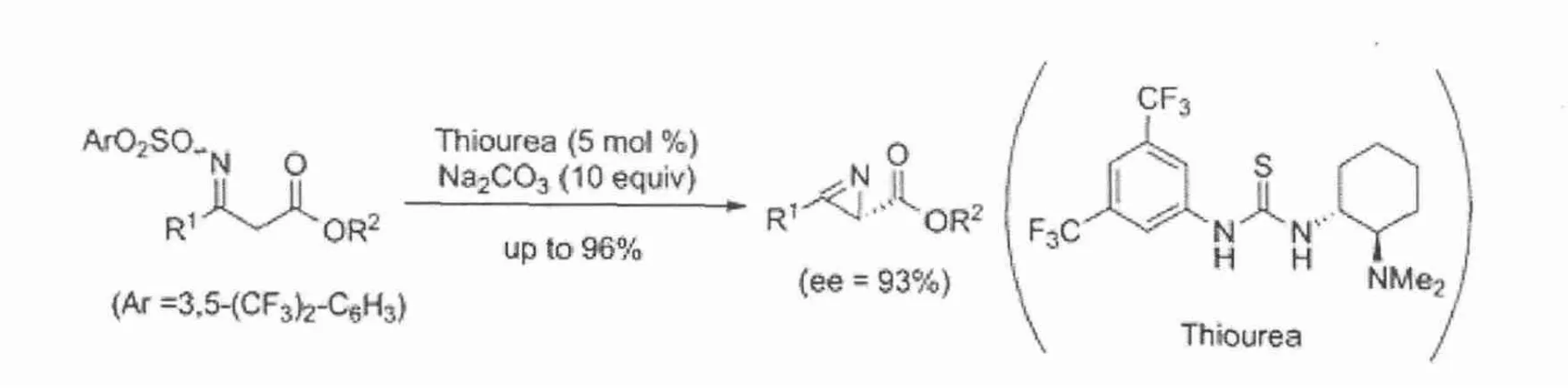
图6 手性硫脲对底物的氢键影响作用[13]
另外,除了常见的中心手性结构硫脲催化剂以外,也有少量对含联萘轴手性结构的手性硫脲催化剂对映选择性催化作用的研究报道[14].该类催化利用联萘本身构造的轴手性环境对底物施加非对称的空间位阻作用,并在双氢键的活化作用下,共同对底物造成手性的催化影响作用.Wang等的研究表明在Michael加成反应中,尽管使用的催化剂的用量只有1%,而所获得的e.e.值最高为97%,产率在78%~92%之间 .这表明中心手性硫脲的不对称催化活性较好之外,轴手性硫脲也具有较高的催化活性,并且双氢键的催化作用模式也可以用于解释轴手性硫脲的不对称催化反应机理.
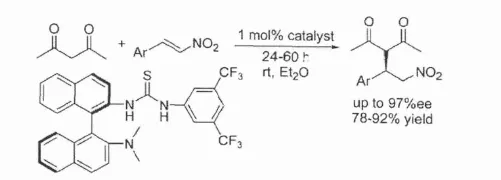
图7 含联萘轴手性硫脲催化Michael加成反应[14]
虽然氢键活化理论较好地解释了手性硫脲的不对称催化反应机理,但MacMillan等[15]提出的SOMO理论则认为一些手性硫脲催化的反应符合手性胺生成烯胺亚胺正离子中间体的机理 .另外,也有一些报道[16]证明,硫脲配体N-H被甲基取代以后,其活性反而有所上升 .这显示了氢键活化理论模型并不能完全涵盖所有手性硫脲的不对称催化反应.

图8 潜手性酮非对称还原反应中单氢键手性硫脲催化剂结构[16]
2 结论
在手性硫脲不对称催化反应研究当中,氢键活化理论无论从实验上,还是从理论计算上都得到了证实,但少量的反应结果并不符合该理论模型 .这说明手性硫脲的不对称催化理论研究仍有待深入 .另一方面,含一个N-H键硫脲手性催化剂仍需要进行更多的研究以探索单氢键理论模型是否同样适用于更多不同结构手性硫脲的不对称催化反应.同时,尽管轴手性有机小分子在Shibasaki类催化剂中显示了良好的对映选择性催化活性,但是,轴手性硫脲的不对称催化反应研究报道相对较少,而构建适当结构的此类手性硫脲催化剂并应用于立体选择性催化反应将是该领域中研究的一个重要方向.
[1]M.S.Sigman,E.N.Jacobsen Schiff base catalysts for the asymmetric strecker reaction identified and optimized from parallel synthetic libraries[J].J.Am.Chem.Soc.1998,120:4901-4902.
[2]林国强,李月明,陈耀全,孙兴文,陈新滋.手性合成-不对称反应及其应用(第四版)[M].北京:科学出版社,2010.57-63.
[3]L.Xian,J.Zhao,M.Chen.Synthesis,Characterization,and Crystal Structure of N-p-Bromophenyl-N’-Phenylacetylthiourea[J].J.Chem.Crystallogr,2009,39:612-614.
[4]P.J.Walsh,M.C.Kozlowski.Fundamentals of Asymmetric Catalysis[M].California:Univ Sci Books,2007.157-160.
[5]张志海,董秀琴,滕怀龙,陶海燕,王春江.含多氢键给体的氨基-硫脲类有机小分子催化剂的设计、合成及应用[J].科学通报,2009,54(22):3407-3419.
[6]W.T.Meng,Y.Zheng,J.Nie,H.Y.Xiong,J.A.Ma.Organocatalytic Asymmetric One-Pot Sequential Conjugate Addition/Dearomative Fluorination:Synthesis of Chiral Fluorinated Isoxazol-5(4H)-ones[J].J.Org.Chem.2013,78:559-567.
[7]Q.Guo,J.C.G.Zhao.Highly Enantioselective Three-Component Direct Mannich Reactions of Unfunctionalized Ketones Catalyzed by Bifunctional Organocatalysts[J].Org.Lett.2013,15:508-511.
[8]H.Xu,S.J.Zuend,M..P Woll,Y.Tao,E.N.Jacobsen.Asymmetric Cooperative Catalysis of Strong BrØnsted Acid-Promoted Reactions Using Chiral Ureas[J].Science,2010,327:986-990.
[9]S.J.Zuend,E.N.Jacobsen.Mechanism of Amido-Thiourea Catalyzed Enantioselective Imine Hydrocyanation:Transition State Stabilization via Multiple Non-Covalent Interactions[J].J.Am.Chem.Soc,2009,131(42):15358-15374.
[10]J.L.Zhu,Y.Zhang,C.Liu,A.M.Zheng,W.Wang.Insights into the Dual Activation Mechanism Involving Bifunctional Cinchona Alkaloid Thiourea Organocatalysts:An NMR and DFT Study[J].J.Org.Chem,2012,77(21):9813-9825.
[11]O.BasleØ,W.Raimondi,M.del M.S.Duque,D.Bonne,T.Constantieux,J.Rodriguez.Highly Diastereo-and Enantioselective Organocatalytic Michael Addition ofα-Ketoamides to Nitroalkenes[J].Org.Lett.2010,12(22):5246-5249.
[12]T.Akiyama,J.Itoh,K.Fuchibe.Recent Progress in Chiral BrØnsted Acid Catalysis[J].Adv.Synth.Catal,2006,348:999-1010.
[13]S.Sakamoto,T.Inokuma,Y.Takemoto.Organocatalytic Asymmetric Neber Reaction for the Synthesis of 2H-Azirine Carboxylic Esters[J].Org.Lett.2011,13(24):6374-6377.
[14]J.Wang,H.Li,W.Duan,L.Zu,W.Wang.Organocatalytic Asymmetric Michael Addition of 2,4-Pentandione to Nitroolefins[J].Org.Lett.2005,7(21):4713-4716.
[15]T.D.Beeson,A.Mastracchio,J.B.Hong,K.Ashton,D.W.C.MacMillan.Enantioselective Organocatalysis Using SOMO Activation[J].Science,2007,27:582-585.
[16]F.Touchard,P.Gamez,F.Fache,M.Lemaire.Chiral thiourea as ligand for the asymmetric reduction of prochiral ketones[J].Tetrahedron Lett.1997,38:2275-2278.
猜你喜欢
分子催化(2022年1期)2022-11-02
高中数理化(2022年14期)2022-08-15
功能材料(2022年5期)2022-06-02
波谱学杂志(2021年3期)2021-09-07
化工管理(2021年7期)2021-05-13
科学与财富(2021年33期)2021-05-10
山西化工(2020年2期)2020-02-16
农药科学与管理(2019年8期)2019-11-23
无机盐工业(2019年6期)2019-06-15
原子与分子物理学报(2019年5期)2019-04-28
