
高效液相色谱-串联质谱法同时测定辽东楤木不同部位中5种皂苷类成分的含量
2014-10-22 12:57:32宋少江刘菲菲
色谱 2014年7期
马 慧,宋少江,刘菲菲,张 艳,彭 缨*
(1.沈阳药科大学药学院,辽宁 沈阳 110016;2.沈阳药科大学中药学院,辽宁 沈阳 110016)
辽东楤木Aralia elata (Miq.)Seem.,五加科(Araliaceae)楤木属(Aralia)植物,其干燥根皮及茎皮味辛、平,有小毒,具有补气安神,强精滋肾,祛风除湿,活血止痛,健胃利水之功效[1]。国内外对其药理作用研究表明:三萜皂苷类成分是楤木发挥药理作用的主要成分,并且具有明显的抗肿瘤活性和提高免疫功能的作用[2-4]。据文献[5-8]报道,目前仅有楤木中单个皂苷(如楤木皂苷A、楤木皂苷C、楤木皂苷Ⅴ、楤木皂苷Ⅵ和楤木皂苷Ⅹ)或总皂苷的测定方法,多采用高效液相色谱-紫外检测法(HPLCUV)和高效液相色谱-蒸发光检测法(HPLCELSD)。但辽东楤木的化学成分复杂,主要有效成分为五环三萜皂苷类,不具有紫外吸收,而且HPLC-ELSD灵敏度低,故采用HPLC-UV和HPLC-ELSD均不能达到同时测定多种微量化学成分的目的。HPLC-MS/MS可以很好地弥补HPLCUV 和 HPLC-ELSD 的不足之处[9,10],不仅能够提供充足的结构确证信息,而且由于其较高的灵敏度和专属性,不需要达到色谱峰的完全分离即可同时准确定量多个化合物,因此在中药质量控制方面受到越来越多的关注[11-13]。
楤木皂苷Ⅱ、楤木皂苷Ⅳ、楤木皂苷Ⅴ、楤木皂苷Ⅹ和楤木叶皂苷Ⅱ为辽东楤木中含量较高的成分,且为辽东楤木叶、种子、芽和根皮中的共有成分,同时这5种皂苷具有明显的药理活性,故本文建立了同时测定楤木皂苷Ⅱ、楤木皂苷Ⅳ、楤木皂苷Ⅴ、楤木皂苷Ⅹ和楤木叶皂苷Ⅱ在楤木药材不同部位中含量的HPLC-MS/MS方法,并考察了药材不同部位之间各皂苷含量的差异,为更合理的开发使用楤木药材提供参考依据。
1 实验部分
1.1 仪器、试剂与材料
API 4000 Q TRAP三重四极杆串联质谱仪(配备ESI源和Analyst 1.6数据采集软件)及Eksigent ultraLC100液相色谱系统(美国Applied Biosystem/MDS SCIEX公司)。
楤木皂苷Ⅱ、楤木皂苷Ⅳ、楤木皂苷Ⅴ、楤木皂苷Ⅹ和楤木叶皂苷Ⅱ对照品由沈阳药科大学中药学院宋少江教授提供,峰面积归一化法测定其质量纯度大于98%,其化学结构式见图1。楤木叶、楤木根皮、楤木芽和楤木种子药材采自辽宁省铁岭市,经沈阳药科大学中药学院宋少江教授鉴别为五加科(Araliaceae)楤木属(Aralia)植物的干燥叶、根皮、芽和种子。
乙醇(色谱纯,山东禹王实业有限公司),乙腈(色谱纯,美国Sigma公司),甲酸(色谱纯,美国Dikma公司),水(天津娃哈哈集团有限公司)。
1.2 溶液配制
对照品储备液:精密称取对照品楤木皂苷Ⅱ3.4 mg、楤木皂苷Ⅴ3.0 mg、楤木皂苷Ⅳ2.6 mg、楤木皂苷Ⅹ3.0 mg和楤木叶皂苷Ⅱ3.2 mg,分别置于5 mL容量瓶中,用70%(v/v,下同)甲醇水溶液溶解并稀释至刻度,摇匀,得对照品溶液。分别精密量取上述对照品溶液1 mL,置于10 mL容量瓶中,用70%甲醇水溶液稀释至刻度,摇匀,得楤木皂苷Ⅱ0.0680 g/L、楤木皂苷Ⅴ0.0600 g/L、楤木皂苷Ⅳ0.0520 g/L、楤木皂苷Ⅹ0.0600 g/L、楤木叶皂苷Ⅱ0.0640 g/L的混合对照品储备液。
供试样品溶液:取不同楤木药材0.1 g,精密称定,置于锥形瓶中,加入70%乙醇溶液100 mL,称定,超声提取60 min,放冷,称定,70%乙醇溶液补足减失的重量,摇匀,过0.45μm微孔滤膜,取续滤液。
1.3 色谱条件
色谱柱为 Alltima C18色谱柱(250 mm×4.6 mm,5μm)。流动相A为0.1%的甲酸水溶液,流动相B为乙腈。梯度洗脱程序为:0~3 min,25%B~30%B;3~18 min,30%B~60%B;18~20 min,60%B~25%B。流速为0.8 mL/min,柱温为室温,进样量为10μL。
1.4 质谱条件
离子源为ESI源;正离子检测方式;源喷雾电压为4.5 kV;离子化温度为650℃;雾化气(GAS1)为0.34 Pa;辅助加热气(GAS2)为0.34 Pa;气帘气(CUR)为0.07 Pa。离子采集方式为多反应监测模式,5种目标组分的其他质谱分析参数见表1。

表1 5种化合物的质谱分析参数Table 1 MS/MS parameters of the five compounds
2 结果与讨论
2.1 质谱条件的优化

图1 (a)楤木皂苷Ⅱ、(b)楤木皂苷Ⅳ、(c)楤木皂苷Ⅴ、(d)楤木皂苷Ⅹ、(e)楤木叶皂苷Ⅱ的化学结构Fig.1 Chemical structures of(a)congmunosideⅡ,(b)congmunosideⅣ,(c)congmunosideⅤ,(d)congmunosideⅩand(e)congmuyenosideⅡ
分别对比了正离子和负离子检测模式。发现在负离子模式下,母离子响应较低,且碎片离子的稳定性较差,其原因可能是由于在负离子模式下离子化程度低;加入甲酸、醋酸铵、氨水后未得到明显改善。而采用正离子模式,[M+Na]+峰响应较好,碎片离子稳定,且加入甲酸后响应强度更高。因此采用正离子MRM检测模式,以不同的碰撞能量打碎待测物的准分子离子,以子离子响应最强时的参数来确定碰撞能量。
2.2 色谱条件的优化
对常用流动相甲醇-水、乙腈-水、乙腈-0.1%甲酸水溶液和乙腈-0.2%甲酸水溶液进行了考察。实验结果表明,使用乙腈-0.1%甲酸水溶液作流动相时灵敏度较高,色谱峰形较好。流动相中不添加甲酸时,色谱峰形较差,色谱峰出现分叉现象,可能是离子化程度较低;加入甲酸使离子化程度大大提高,峰形明显改善;但是加入0.2%的甲酸时,灵敏度稍有下降,说明流动相中甲酸的含量对质谱的响应也有影响。最终确定采用乙腈-0.1%甲酸水溶液作为流动相进行梯度洗脱。待测组分的色谱图见图2。
2.3 线性关系及检出限
取各对照品储备液适量,精确配制不同质量浓度的系列混合标准溶液,于优化后的条件下分别进样测定。以各物质特征碎片离子(定量离子)的峰面积(y)对被测组分的质量浓度(x,μg/L)进行线性回归,得到回归方程、相关系数(r2)和线性范围;以信噪比(S/N)为3确定5种待测组分的检出限;结果见表2。

图2 对照品溶液和供试品溶液的LC-MS/MS色谱图Fig.2 LC-MS/MS chromatograms of the reference solution and sample solution

表2 5种组分的线性方程、相关系数、线性范围及检出限Table 2 Calibration equations,correlation coefficients(r2),linear ranges and detection limits of the five compounds
2.4 精密度和稳定性
精密吸取1.2节的对照品储备液适量,制成合适浓度的混合对照品溶液,在优化的实验条件下测定,重复进样6次,每次10μL,测定峰面积。结果表明楤木皂苷Ⅱ、楤木皂苷Ⅳ、楤木皂苷Ⅴ、楤木皂苷Ⅹ和楤木叶皂苷Ⅱ峰面积的RSD分别为2.0%、2.5%、2.7%、2.4%和2.5%,均符合要求。
取楤木叶干燥粉末,按照1.2节方法制成供试品溶液。分别在制备好的0、4、8、12、24 h进样,按1.3节的色谱条件测定峰面积。结果楤木皂苷Ⅱ、楤木皂苷Ⅳ、楤木皂苷Ⅴ、楤木皂苷Ⅹ和楤木叶皂苷Ⅱ 的 RSD 分 别 为 1.5%、1.7%、1.6%、0.9% 和1.5%。结果表明,供试品溶液在24 h内稳定。
2.5 加样回收率
精密称取已知这5种皂苷成分含量的楤木叶粉末6份,每份0.1 g,精密加入适量的对照品溶液,按照1.2节方法制成供试品溶液,按优化后的条件测定,结果均符合要求(见表3)。
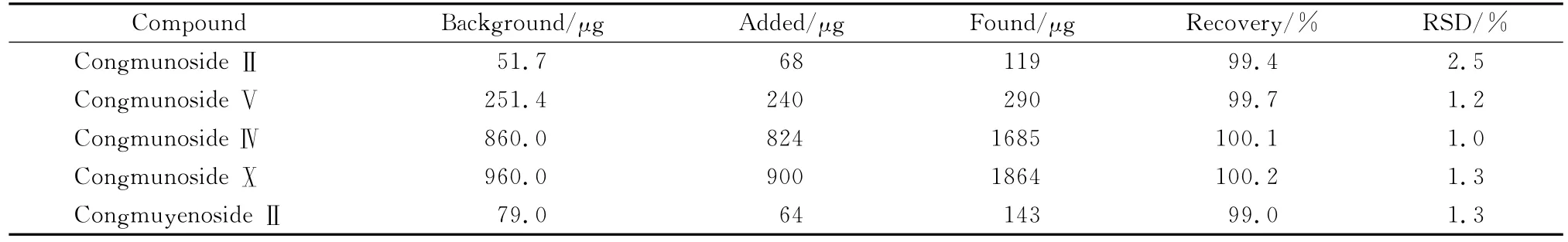
表3 楤木叶中5种三萜皂苷成分的加标回收率(n=6)Table 3 Spiked recoveries of the five triterpenoid saponins in the leaves of Aralia elata(n=6)
2.6 样品测定
取不同部位的楤木适量,按1.2节供试品制备方法操作,按优化后的条件测定峰面积并计算含量,结果见表4。

表4 楤木不同部位中的5种三萜皂苷类成分的含量(n=3)Table 4 Contents of the five triterpenoid saponins in different parts of Aralia elata(n=3)
测定结果表明,上述5种皂苷总含量的多少顺序为根皮>叶>种子>芽,证明楤木根皮作为药用部位具有一定的合理性。楤木叶中被测5种皂苷的量也较高,提示楤木叶中也含有大量的皂苷,而楤木芽及楤木种子中5种皂苷含量相对较低。这一结果可以为对楤木中的皂苷进一步研究以及药材部位的选择提供参考。
3 结论
本文建立了高效液相色谱-串联质谱同时测定楤木皂苷Ⅱ、楤木皂苷Ⅴ、楤木皂苷Ⅳ、楤木皂苷Ⅹ和楤木叶皂苷Ⅱ含量的方法。实验结果表明,该方法具有简便、快捷、准确、灵敏的特点,可用于测定辽东楤木药材不同部位中的皂苷类成分的含量,进而为楤木药材的质量控制和药物疗效、药代动力学等相关研究提供新的参考。
[1]Sun G B,Wang M,Gao M M,et al.Chinese Pharmacological Bulletin(孙桂波,王敏,高蒙蒙,等.中国药理学通报),2013,29(6):773
[2]Li R,Zhang X,Ren M P,et al.Lishizhen Medicine and Materia Medica Research(李蓉,张潇,任美萍,等.时珍国医国药),2011,22(9):2199
[3]Wang C M,Zhang G M.Acta Chinese Medicine and Pharmacology(王春梅,张广美.中医药学报),2011,39(1):17
[4]Zhang Y,Peng Y,Li L Z,et al.Food Chem,2013,83(4):806
[5]Wang Q H,Wang Q,Wang Z B,et al.Chinese Traditional Patent Medicine(王秋红,王琦,王知斌,等.中成药),2013,35(9):1950
[6]Song S J,Li L,Ma Z Q,et al.Journal of Shenyang Pharmaceutical University(宋少江,李丽,马志强,等.沈阳药科大学学报),2006,23(3):162
[7]Liu Y,Tian J,He B,et al.West China Journal of Pharmaceutical Sciences(刘艳,田吉,何兵,等.华西药学杂志),2010,25(2):217
[8]Wu R F,Zhao S Y,Liu Y J,et al.Chinese Journal of Basic Medicine in Traditional Chinese Medicine(吴睿凡,赵松岩,刘运嘉,等.中国中医基础医学杂志),2012,18(10):1136
[9]Zhao L L,Liu F F,Peng Y,et al.Chinese Journal of Chromatography(赵璐璐,刘菲菲,彭缨,等.色谱),2012,30(12):1271
[10]Zhang J C,Wei J,Zhong H M,et al.Chinese Journal of Chromatography(张敬彩,魏杰,钟虹敏,等.色谱),2013,31(1):79
[11]Chi Y M,Li Y,Zhang Y,et al.Chinese Journal of Chromatography(池玉梅,李瑶,张瑜,等.色谱),2013,31(9):838
[12]LüC,Ding T,Ma X,et al.Chinese Journal of Chromatography(吕辰,丁涛,马昕,等.色谱),2013,31(11):1046
[13]Qin S,Wang J,Xu Y J.Chinese Journal of Chromatography(覃莎,王锦,徐远金.色谱),2012,30(11):1153
猜你喜欢
昆明医科大学学报(2021年4期)2021-07-23 01:21:36
儿童绘本(2019年21期)2019-12-06 07:38:12
中成药(2018年9期)2018-10-09 07:19:04
中国蜂业(2018年4期)2018-05-09 06:25:08
中成药(2017年9期)2017-12-19 13:34:40
中成药(2017年6期)2017-06-13 07:30:34
当代化工研究(2016年6期)2016-03-20 16:21:46
意林(2014年10期)2014-07-28 18:37:04
中国药业(2014年16期)2014-05-14 06:46:20
无机化学学报(2014年3期)2014-02-28 17:30:58
