
三氟甲烷(HFC-23)的资源化转化利用
2014-10-13 07:58韩文锋靳碧波周强王树华
化工进展 2014年2期
韩文锋,靳碧波,周强,王树华
(1浙江工业大学工业催化研究所,浙江 杭州 310014;2巨化集团研究院,浙江 衢州 324004)
三氟甲烷(CHF3,HFC-23,氟仿,R-23,FE-23)是R-22(CHClF2)生产过程中产生的一种不可避免的副产物。目前,它作为灭火剂或刻蚀剂等仅有少量应用。然而,CHF3的全球温室效应潜值(GWP)是CO2的14 800倍,在大气层的寿命高达264年,且是目前已知的温室效应第二高的温室气体,仅次于 SF6[1]。
CHF3主要是在 CHClF2(R22,HCFC-22) 生产过程中产生的[2]。按现有技术,生产1 t的CHClF2会产生大约40 kg的CHF3副产物。根据估算,在2015年 CHF3的排放量将达 2.4万吨,相当于 2.8亿吨的CO2排放[3]。CHClF2曾是应用最为广泛的制冷剂及推进剂之一。由于它是臭氧层破坏物质,自1987年《蒙特利尔议定书》及后续的《京都议定书》签订后,它的应用逐渐减少。但是,CHClF2是生产诸如 TFE(四氟乙烯)、PTFE(聚四氟乙烯)及其他氢氟烃(HFCs)的基本原料。由于这些应用不可避免,用于四氟乙烯、聚四氟乙烯等的生产而使用CHClF2被排除在议定书规定的减排条目外。因此,CHF3的排放仍将长期持续。通常,在五氯化锑的催化作用下,CHClF2的合成包括式(1)~式(3)三个反应。
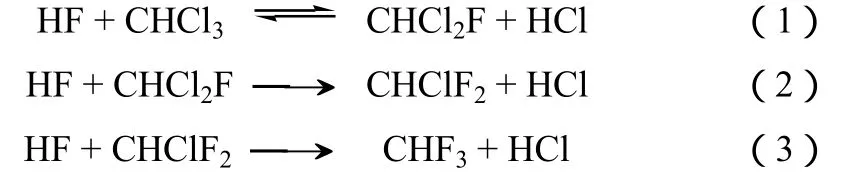
虽然通过工艺优化CHF3的量可以大幅度降低,但是由于热力学限制,CHF3的产生仍不可避免。此外,工艺优化往往伴随着装置产量和经济效益的降低。根据模型计算,经过优化的装置其副产量可以从3%~5%降低到1.5%~3%[4]。
目前,CHF3的减排方式主要是通过联合国环境规划署(UNEP)负责的清洁发展机制(CDM)项目,采用高温焚烧(1200 ℃)进行的。通过CDM项目,发达国家出资向发展中国家购买温室气体减排指标。根据联合国环境规划署(UNEP)的统计数据,我国 CHF3的排放量占全世界排放量的一半以上,在CDM项目中通过焚烧CHF3将减排相当于8260万吨的CO2。 碳交易目前的市场价格为2.81欧元/t,8260万吨的 CO2为国家带来相当于约为19.72亿人民币的经济效益。同时按照当前趋势估算,在2050年,含氟烃的排放(CO2当量)将占全球温室气体排放的 9%~19%,故若将这些废气如CHF3催化转化和资源化利用将带来巨大的经济和能量效益。
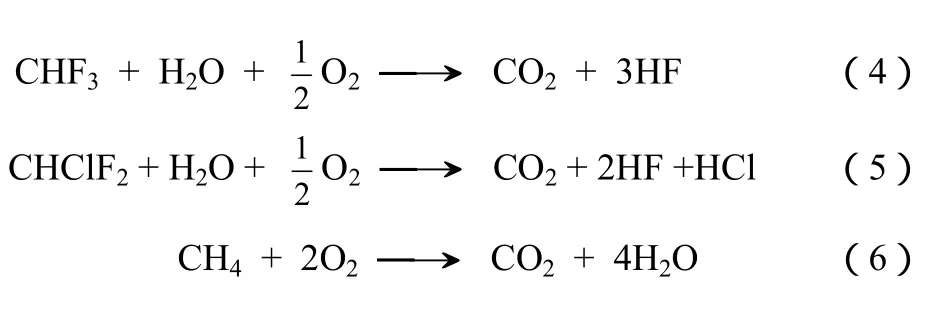
目前可采用的 CHF3的处理方法主要包括热焚烧、等离子处理等。热焚烧是经过联合国环境署审定并在 CDM 项目中采用的路线[5]。在该路线中,CHF3通过与液化石油气在 1200 ℃燃烧,经过式(4)~式(6)三个反应转化为CO2、HF等。
根据联合国全球气候变化框架协议组织(UNFCCC)的数据,目前约有20个CHF3焚烧项目进入CDM进行碳减排交易。
等离子处理是另一个已经商业化运行的工业处理路线。1992年,澳大利亚联邦科工组织(CSIRO)和 SRL Plasma Ltd. 首先联合开发了所谓的PLASCON工艺用于氟利昂(CFCs)的处理,并在2007年被用于其他ODS和CHF3的处理[6-7]。
由于此类物质性质稳定,且可以作为灭火剂,因此采用热裂解或焚烧处理往往需要极为苛刻的反应条件,处理的成本和设备投资往往较高。等离子体工艺的设备投资和运行成本是一般热焚烧路线的3~5倍,也不是最佳选择。如CHF3,即使在燃烧条件下分解也较为困难。从而诸如焚烧和等离子处理等消除法在能量上来看是极为不利的。此外,CHF3拥有宝贵的C-F键,利用它来合成环境友好、附加值高的其他含氟化合物是更为理想的路线。值得一提的是,正如前面所述,CHF3是在CHClF2生产过程中形成的唯一含碳副产物(另一个不含碳副产物为HCl),因此得到的CHF3废气纯度往往高于其他一般工业废气[8]。这使得CHF3更有利于成为其他含氟化合物合成的原料。
1 CHF3裂解转化为C2F4(TFE,四氟乙烯)和C3F6(HFP,六氟丙烯)
四氟乙烯(TFE)是涂料、黏结剂和特富龙(Teflon,PTFE)等工业产品的主要生产原料之一。在工业生产过程中,四氟乙烯主要通过 CHClF2(R22,HCFC-22)在600~900 ℃气相裂解得到,经过条件优化其得率可以超过95%[9]。其主要的反应见式(7)和式(8)[10]。
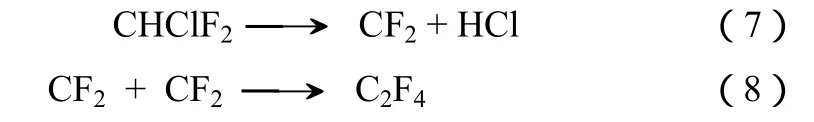
目前有关CHF3裂解制TFE的研究相对较少。在更高的温度下(>750 ℃),CHF3生成TFE具有和HCFC-22完全类似的反应机理,见式(9)[11-13]。

其分解动力学可以表示为

显然,CHF3分解是一级反应。也就是说,在高温下,CHF3直接脱去HF形成CF2。在较低的压力下(压力低于0.29 atm),该反应也可能遵循二级反应机理[12,14],其反应机理可以表示为式(10)~式(13)。

在低压下,CHF3首先通过与反应体系里的其他物质如N2、反应器壁[式(10)里的M]碰撞获得能量,形成活化态的 CHF3*后分解。由于在 CHF3裂解反应的实验中并未检测到痕量的C2F5H,因此二级反应机理[式(12)]被证实不是CHF3分解的主要通道[10]。
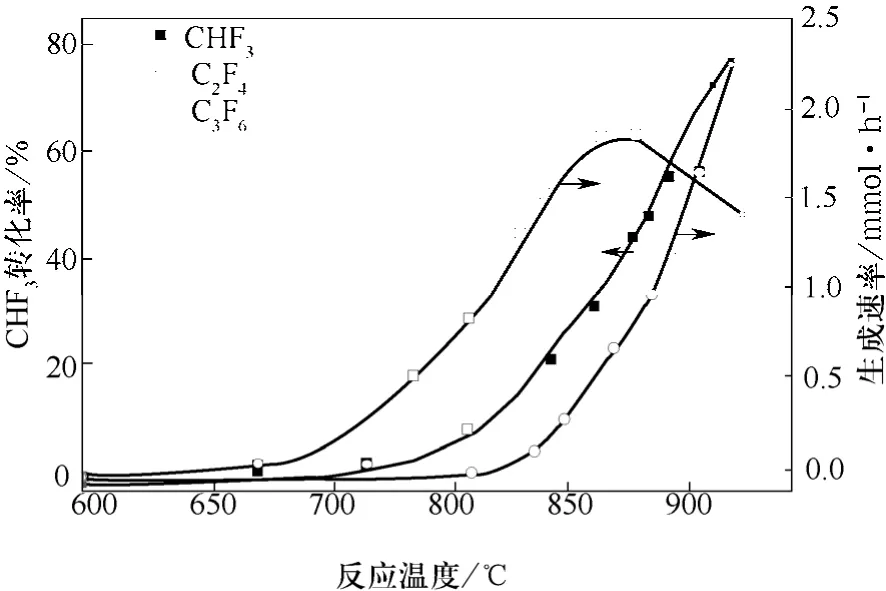
图1 CHF3和N2的进料比为1∶9,且在常压下温度对CHF3气相分解的转化率和C2F4以及C3F6的生成速率的影响[12](停留时间:0.5 s)
图1是在温度范围为600~900 ℃、反应停留时间为0.5 s及常压下,CHF3用9倍N2稀释后的裂解结果[15]。在研究的条件下,得到的主要产物是TFE和HFP(六氟丙烯)。在反应温度为850 ℃时,TFE生成速率最高,随着温度继续升高,其产率持续下降。HFP的生成速率随着反应温度的升高而逐步升高。在860 ℃下,TFE和HFP的选择性分别为31%和16%,而在温度升至900 ℃后,它们的选择性分别变为17%和41%。
当 CHF3和 TFE一起通入反应管进行共裂解时,HFP的产率可以更高。表1是CHF3和TFE在温度为870~880 ℃、反应停留时间2 s时的共裂解结果[16-17]。在此反应条件下,反应的主要产物为HFP。正如反应式(8)所示,在高温条件下,CHF3脱HF生成CF2自由基。CF2继续和C2F4反应形成C3F6,如式(14)~式(17)。
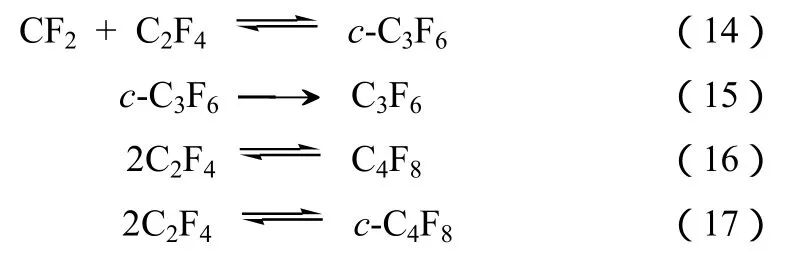

表1 不同进料比对R23和TFE气相热解反应的影响
杜邦公司研究人员则利用CHF3和CHClF2反应混合物在625~800 ℃ 的温度和少于2 s的接触时间下进行裂解,最终得到了包括 TFE、HFP、五氟乙烷及七氟丙烷等主要产物[18]。令人意外的是,热解反应中,CHClF2的存在显著降低了CHF3的分解温度。他们认为,反应中三氟甲烷的作用是增加饱和的 C2及 C3化合物如 CF3CHF2(HFC-125)及CF3CHFCF3(HFC-227ea)的量,但具体机理还不清楚。研究发现,KF/AC(AC指活性炭)催化剂能够显著催化 CHF3的高温裂解。在相同条件下,催化剂能使TFE和HFP的收率提高数倍以上,结果见表2。
由表2可知,在KF/AC的催化下,CHF3的转化率提高了近4倍,TFE和HFP的收率提高了近2倍和10倍。如前所述,在无催化剂存在下,CHF3在高温下分解脱HF形成CF2自由基,然后两个CF2自由基聚合形成 TFE(该反应活化能接近于零)。而要使生成的TFE和CF2继续反应生成HFP能垒相对较高(51 kJ/mol),指前因子相对较低[19]。据推测,在活性炭催化剂存在下,CHF3分解形成的CF2强烈吸附于AC表面 ,从而有利于其自身的再次聚合,形成TFE及HFP,降低了反应能垒。
图2是CHF3在KF/AC催化剂上裂解产物TFE和HFP的得率随反应温度升高的变化趋势。和图1的无催化气相裂解类似,HFP的得率随着温度的升高而逐步提高,而TFE则随温度升高达到一个最高之后逐步下降。如表1所示,优化反应后,在872 ℃时CHF3和TFE共裂解,HFP的选择性为28.5%。而即使用CHClF2作为原料,在金属氟化物催化下,反应条件为常压、650 ℃、空速15 000 h−1时,TFE的收率也仅为28.7%[21]。如图2所示,在类似的反应条件下 (840 ℃),仅利用 CHF3单组分裂解,KF/AC催化剂HFP的得率就高达20.2%。在800 ℃,TFE和HFP的总收率接近35%。

图2 温度对CHF3在KF/AC催化剂上催化裂解生成TFE和HFP收率的影响[20](空速:4300 h−1)
2 CHF3和 CH4反应合成 VDF(偏氟乙烯,CH2=CF2)
VDF是合成PVDF(聚偏氟乙烯)等具有特殊物理化学性质聚合物的基本单体之一[22]。这些聚合物被广泛应用于航空、电子、化工、油田、半导体等各种行业,具有长寿命、可靠、免维护、适应恶劣环境等特点[23]。有关含 VDF的聚合物的综述可见参考文献[24]。
工业化大规模 VDF合成主要是通过氢氟氯烃在300~1000 ℃气相裂解进行的,其主要的反应如式(18)~式(21)。
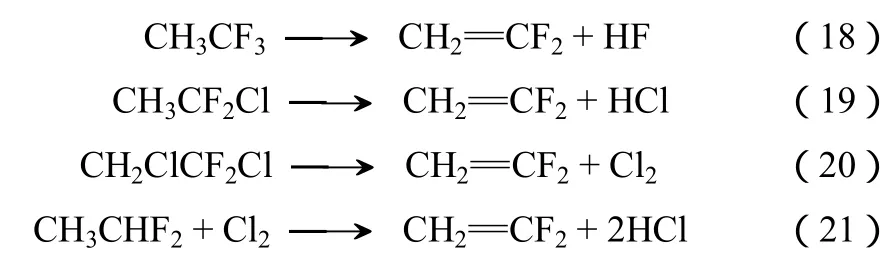
CBrClF2(哈龙 1211)、CCl2F2(CFC-12)等曾被广泛用作灭火剂、制冷剂、发泡剂。由于他们是主要的臭氧层破坏物质(ODS),在《蒙特利尔议定书》及后续的《京都议定书》等规定中,这些物质将被逐步废弃并禁止生产。研究发现,以废弃物质资源化利用为目标,这些物质与CH4进行高温反应后能合成环境友好、附加值高的化学品VDF[25-30]。以哈龙1211和CFC-12为例,其反应机理如式(22)~式(30)。

目标产物VDF通过反应式(30)由CH3和CF2自由基聚合而成[31]。CHClF2与甲烷的反应也同样可以合成VDF。在反应温度高于725 ℃,反应式(22)可以实现100%转化,而在温度高于800 ℃时,VDF的收率可达43%以上。反应的主要产物TFE和VDF的产率随温度的变化如图3[10]。
采用CFCs、HFCs和HCFCs的混合废气(回收的废弃制冷剂,其主要组成为 CHClF2、CCl2F2和 CH2FCF3,比例为9∶8∶1),利用相同的反应路线也可以达到类似的资源化利用的目的[32]。

表2 KF/AC催化剂和无催化剂对分解CHF3的影响[20](反应温度800 ℃)

图3 N2、CH4和反应式(23)的进料比为 10∶1∶1和常压下,不同温度对反应式(22)和CH4热解反应的影响[10](停留时间为0.5s)。
同样,本工艺路线也适用于 HFCs如 CHF3与甲烷反应合成 VDF[33]。在反应温度为 900 ℃时,CHF3的转化率为77%,VDF的选择性为27%,收率高达21%[15]。
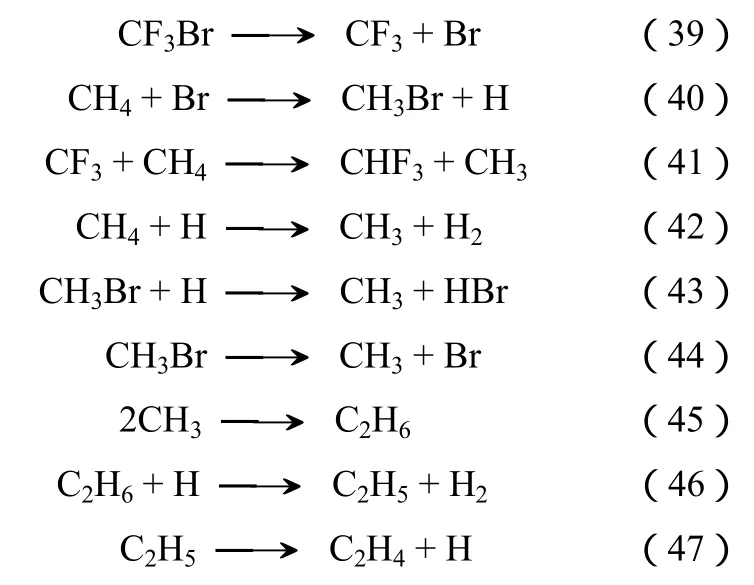
图4是等量的CHF3和CH4反应的结果,主要的产物是 VDF、TFE及 HF,副产物包括 C3F6、CH2F2、C2H3F、C2HF3、C2H6、C2H2和 CHF2CHF2。痕量的产物有 CF3CH=CF2、CH2=CFCF3、C4H2F2、C3H8和CHF=CHF。CHF3和CH4的转化率随反应温度升高而逐渐提高,但在研究范围内,CH4的转化率远低于CHF3。在低温下,TFE和VDF的生成速率较为接近,但在高温条件下(温度高于840 ℃),VDF逐步变为决定性的产物。
不同于哈龙1211和CFC-12,CHF3除了C—F而不含有C—Br和C—Cl。C—F是已知的最为稳定的化学键之一,因此在CH4和CHF3反应体系中,缺乏诸如Br、Cl、F等自由基来活化甲烷。CH4极为稳定,即使在900 ℃的高温,也仍然没有发现大量的分解[34]。前面提到,VDF主要通过CH3和CF2耦合而成[31],显然CH3来自于CH4,而CF2来自于CHF3脱HF裂解。由于如图4所示的CH4转化率较低,因此反应体系的CH3不足,造成部分CF2未能与CH3耦合,而是发生自身聚合形成TFE。

图4 N2、CHF3和CH4的进料比为 10∶1∶1和常压下,温度对CHF3和CH4热解反应的影响(停留时间为0.5 s)
CF2优先和 CH3耦合形成 VDF的原因可以从CF2自身聚合[反应式(8)]和 CH3与 CF2[反应式(30)]耦合的各自的反应速率常数得到解释。虽然这两个反应的活化能均接近于零,但是反应式(30)的反应速率常数的指前因子是反应式(8)的10倍,这意味着在同等条件下反应式(30)的反应速率是反应式(8)的10倍左右。由于在反应条件下CH4自身不能分解,根据机理分析,图4中甲烷的转化率是通过一系列的链式反应达到的。
引发

链传递


此体系中链是由发生于反应管壁的类似于催化反应的表面反应引发的[35]。很显然,在加入少量气相自由基引发剂如CBrF3及CH3OH后,VDF的收率可以进一步提高。
在少量的 CBrF3存在下(6000 μL/L),CH4和CHF3反应中CH4的转化率提高了1~4倍,结果见图5。相比之下,少量CBrF3的引入对CHF3的转化率几乎没有影响。在未添加 CBrF3的情况下,CH4活化涉及的一系列链式反应由发生于管壁的表面反应引发,效率较低。加入CBrF3后,由于发生反应(39)~反应(47)的一系列反应,CH4的活化被明显加速,从而显著地提高了CH4转化率。相应地,由于反应体系中大量的CH4分解,CH3自由基的浓度大幅度提高,因而CH3与CHF3分解得到的CF2自由基耦合的概率提高,从而使得VDF的收率大大提高。在未添加CBrF3时,TFE是反应的一个主要副产物,在添加少量CBrF3后,只有微量的TFE被检测到[36]。
在未添加CBrF3且反应温度为850 ℃时,CHF3与CH4反应生成VDF的选择性和收率分别为23%和8.1%;当反应温度提高到900 ℃时,CHF3的转化率提高为77%,VDF的选择性提高为27%,且收率达到21%[15]。当加入少量CBrF3而反应温度仍为850 ℃时,VDF选择性和收率分别提升至 55%和16%,提高了一倍以上;温度提升至900 ℃时,VDF的选择性和收率更是达到37%和26.6%[36]。
此外,加入少量CH3OH[37]、CaBr2[38]等促进剂(图6 )也能显著提高VDF的收率,但是其收率仍远远低于 CHClF2与 CH4反应[10,37]或 CH3Br与CHClF2反应所得的收率[29]。值得一提的是,CH3OH和CaBr2的作用机理不同于CBrF3,在反应温度下,CH3OH易分解为H、OH、HCHO及CH2OH等自由基,促进CH4和CHF3的活化,从而提高转化率和目标产物的收率。研究发现,CaBr2的作用机理是CHF3首先和CaBr2作用选择性地形成CBrF3,然后CBrF3再作用于CHF3与CH4的反应。
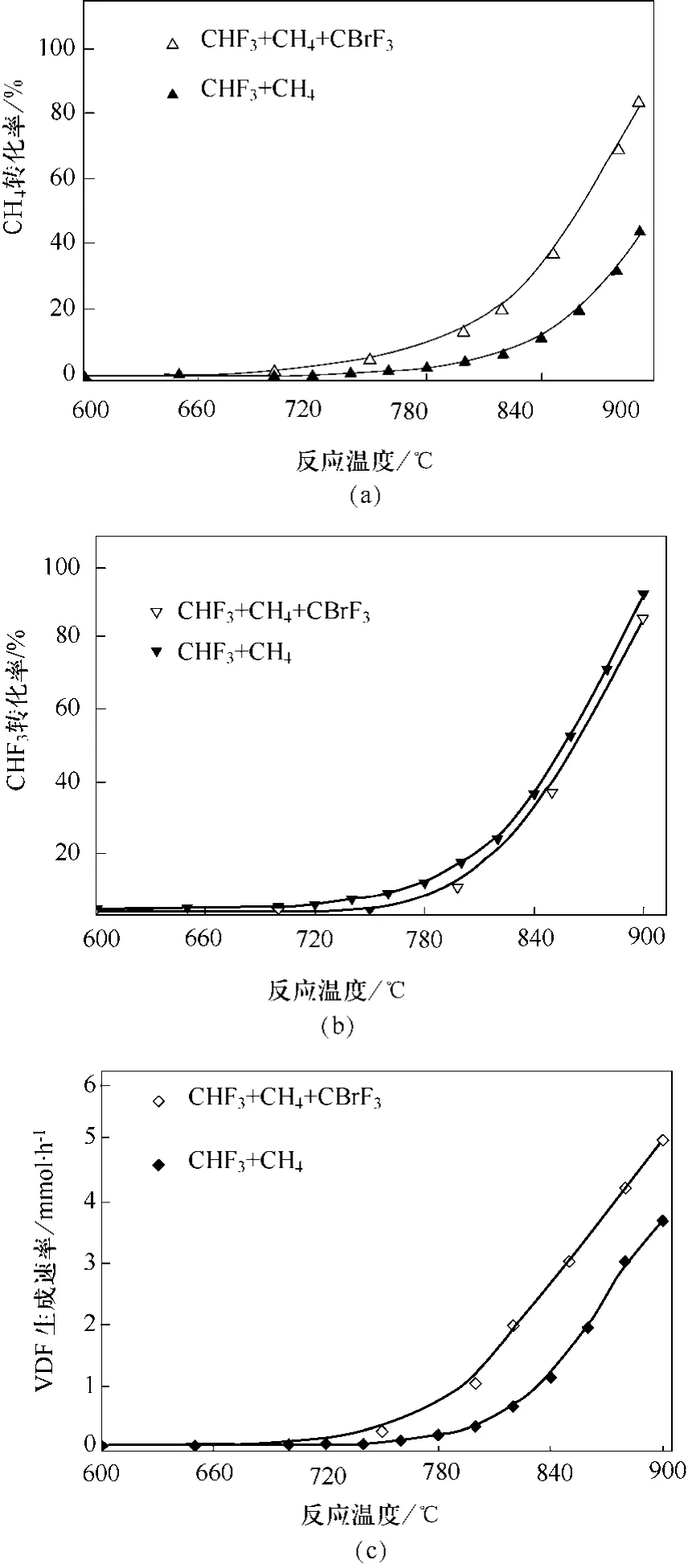
图5 N2、CHF3和CH4的进料比为 10∶1∶1和常压下,添加少量 CBrF3对CHF3 和CH4 热解反应的影响[36](停留时间为0.5 s)
3 CHF3和I2反应合成CF3I

图6 促进剂对CHF3和CH4生成VDF的影响[35](空速为0.5s)
在《蒙特利尔公约》和《京都议定书》所规定的控制措施下,哈龙 1301(三氟溴甲烷,CBrF3)作为主要的破坏臭氧层受控物质之一,除一些特殊场合如军事设施外,其作为灭火剂、发泡剂和制冷剂等应用被严格禁止。我国也签署了京都议定书,按照承诺,在2010年停止使用哈龙产品。与FM-200(CF3CHFCF3)和 CHF3等替代品相比,三氟碘甲烷(CF3I)与哈龙1301性能最为接近。此外,其温室效应值(GWP)仅为5,相比CHF3的14800几乎可以忽略不计[39]。早在 1992年,该物质通过美国国家消防规范(NFPA)认证,被批准使用。
三氟碘甲烷的灭火性能与哈龙1301基本类似,且同为气体灭火剂,灭火后不留痕迹,其单位质量灭火效率高,存储体积小,是一种理想的哈龙类产品的替代品[41]。早期三氟碘甲烷通过三氟乙酸盐与I2的均相反应得到,收率可以达到60%~80%[42]。然而,此路线成本较高,且不适合连续化生产。
日本学者Nagasaki等[40]首先发现K负载的活性炭催化剂能够催化CHF3、O2和I2的气相反应,合成CF3I,从而实现其连续化生产。此外,其他碱金属和不同钾盐对合成 CF3I的也具有不同的催化效果[40],见表3。
如表3所示,在反应温度为550 ℃、空速为30 h−1时,CF3I的收率最高可以达到 39%。但由于反应空速较低,工业生产时产量也较低。但是如果将空速提高至60 h−1,CF3I的收率也随之降至16%。
通过实验Nagasaki等[40]提出了如图7所示的反应机理。不同于无催化剂的气相高温裂解反应,KF/AC在500 ℃就能显著促进CHF3的分解并生成CF2。在无催化剂的情况下,如前所述,CF2自由基自身耦合形成TFE和HFP。而在催化剂作用下,认为CF2首先强吸附于活性炭表面,从而不易发生自身聚合,而是进行自身歧化反应,生成C和CF3。CF3再与I2作用最终生成CF3I。因此,根据此机理的反应如式(48)。
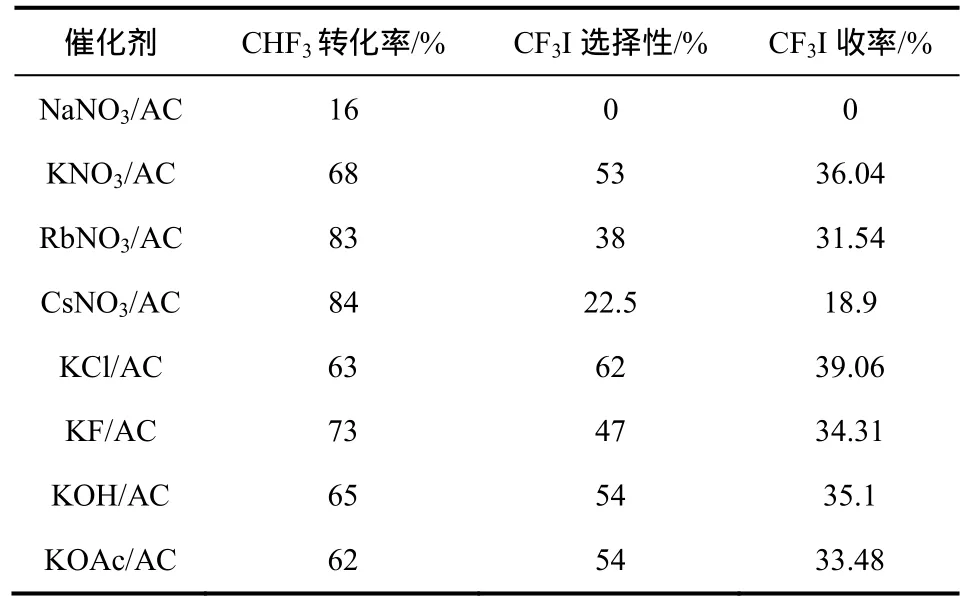
表3 碱金属催化剂和碱土金属催化剂对合成CF3I的影响①[40]

图7 CHF3和I2在KF/AC 催化剂上合成CF3I的反应机理[40]

即使CHF3100%选择性地分解为CF2,3个CF2自由基也只能生成2个CF3,因此该反应的CF3I理论选择性也不会超过67%[43]。正因为此反应形成大量的积炭,因此催化剂在几个小时内就失活。在反应体系引入O2,烧除积炭,能明显改善催化剂寿命。但由于活性炭载体对氧气不稳定,反应一段时间后比表面积降低,催化剂活性组分明显流失[44]。
在活性炭催化剂上,Yang等[45]的研究也发现CHF3在其表面催化分解形成的CF2发生歧化反应,形成CF3和C,与Nagasaki提出的机理一致。但是在另外的实验中,他们检测到了大量的CF4及C2F6等产物,因此推测CF2在活性炭上也可以进行图8所示的反应[46]。
吸附于活性炭上的CF2直接分解为C和F,然后F与CF3结合形成CF4产物。但是,本反应需要直接打断2个C—F,能垒较高,因而可能性较低。Han等[20]研究认为,活性炭能直接催化CHF3分解为CF3,CF3在活性炭表面歧化形成CF4等产物。更确切和详细的反应机理仍有待于更为深入的实验研究。
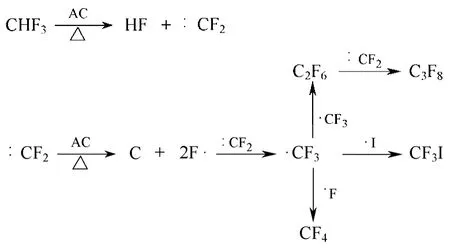
图8 CF2在活性炭上的歧化反应机理[46]
4 CHF3作为三氟甲基化原料
三氟甲基化反应是制药及其他有机化合物中间体合成的重要反应过程之一[47]。通常,三氟甲基化的原料包括CF3I、CF3Br、CF3SiR3(R指有机烷基基团)、CF3COOX、CF3SO2Cl等,成本均较高[48]。
CHF3是较有吸引力的三氟甲基化试剂。其作为原料具有易得、价廉、反应具有原子经济性、无毒及不破坏臭氧层等特点。作为一种气体,CHF3可以溶解于许多有机溶剂。但是,其作为一种极弱的有机酸(pKa = 27,酸性远远弱于氯仿),要使其活化往往需要极强的碱(如二甲基亚砜基钾、叔丁醇钾)来脱质子[48]。
长期以来,研究者们期望直接利用 CHF3作为三氟甲基化试剂,虽做了很多尝试但很少获得成功。然而利用多步法将 CHF3转化为其他化合物,从而间接地合成CuCF3却有不少报道[48]。例如,利用叔丁醇钾与 CHF3作用合成苯基砜类或亚砜类三氟甲基类化合物(图9),在−50 ℃的反应条件下,再利用它们与CuI作用形成CuCF3。CuCF3再与碘苯类化合物反应就可得到三氟甲基化的苯类化合物。
最近,有研究发现[49],在2t-BuOK (2-叔丁醇钾)作用下(图10),CuCl能和CHF3在常温常压下形成CuCF3,而不需要超低温(如前面的−50 ℃,防止 CF3过度分解)。利用特定的稳定剂,得到的CuCF3可以和 PhB(OH)2类化合物反应得到一系列的三氟甲基化芳烃[50]。

图9 多步合成三氟甲基苯[48]

图10 2t-BuOK 和 CuCl 的存在下直接合成三氟甲基化芳烃[49]
几乎在同时,《Science》期刊报道了更为简便的利用 CHF3作为三氟甲基化试剂的方法[51]。将CHF3溶解于四氢呋喃溶液中,在−40 ℃利用等量的六甲基二硅烷重氮钾 (KHMDS) 作为碱性化合物活化 CHF3,与三甲基硅氯化物(TMSCl)反应形成三甲基硅氟化物(TMSF)。利用 TMSF和Si、B、S及C基化合物反应可以得到各种三氟甲基化化合物,见表4。正如德国氟化学家Haufe所说,这使得原本无用的温室气体“重回工作岗位”(“Putting a Greenhouse Gas to Work”),它提供了一种具有原子经济性和环境友好的三氟甲基化路线[52],见图11。

图11 六甲基二硅烷重氮钾和三甲基硅氯化物的存在下直接合成三氟甲基硅氟化物[51]
5 结 语
目前,由于温室气体的排放引起的环境问题日益受到关注,关于温室气体的研究和讨论主要集中于CO2的排放上。事实上,虽然CHF3的排放量相对较少,但由于其具有极高的温室效应潜值(是CO2的14 800倍),其对温室气体的减排效应不可忽视。相对于CO2,目前有关CHF3的减排研究极少。
不同于其他废弃物,CHF3往往具有较高的纯度,且其含有宝贵的C—F键,利用高温或等离子进行销毁从能量和资源利用角度来说均不是最佳选择。受惠于联合国CDM项目的碳交易计划,利用CHF3焚烧进行温室气体减排,我国可以获得一定的碳交易补贴。但是,由于经济原因,碳交易价格逐年下跌,加上其他不稳定因素,依靠国外碳交易补贴进行 CHF3焚烧减排温室气体难以长期维系。因此,尽早开展 CHF3减排,特别是其资源化利用是十分迫切和必要的。
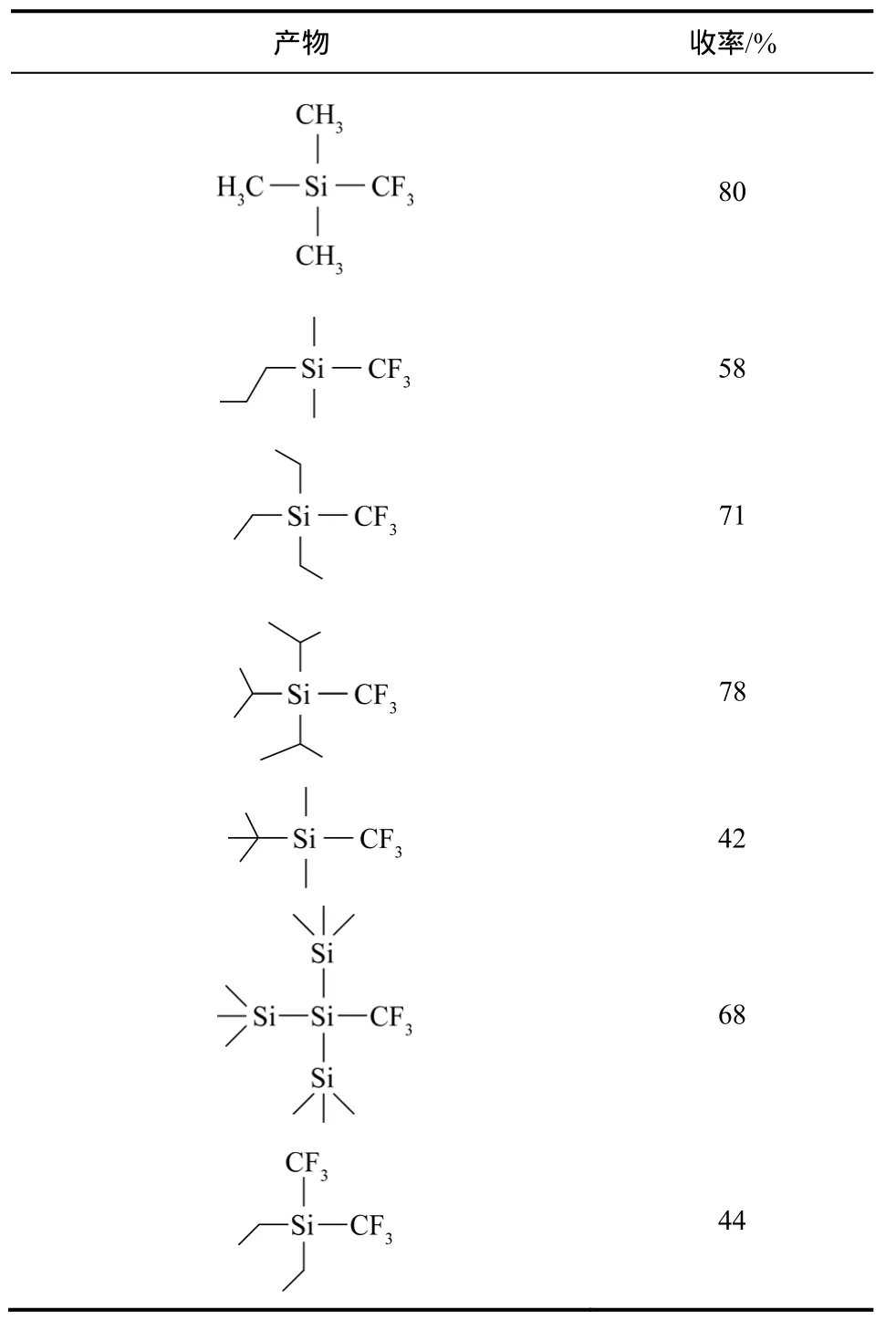
表4 CHF3与不同三烷基硅氯化物反应得到的三氟甲基化产物及其收率
由于 CHF3的排放较为集中,纯度较高,因此对其实施资源化利用是十分有利的。研究已经发现,CHF3可以转化为附加值高、环境友好的TFE、HFP、VDF、CF3I等产物。它也可以作为三氟甲基化试剂,用于制药及其他有机中间体的合成。虽然利用CHF3裂解合成TFE及HFP需要的温度相对较高,收率偏低,难以与现有的反应式(23)工业路线竞争,但是在适合的催化剂作用下,反应温度可以大幅度降低,TFE和HFP的收率可以明显提高。因此该路线的关键问题是合适的催化剂的开发。利用 CHF3合成VDF是另一条较有吸引力的路线。VDF附加值高于 TFE,且其通过与另一个廉价的反应物(CH4)反应得到。利用其他助剂,VDF的选择性和收率可以明显提高,具有一定的工业价值,其缺点是副产物较多。而利用CHF3与I2反应合成CF3I已由日本的TOSOH公司实现工业化中试生产。虽然CF3I作为灭火剂及制冷剂的替代品的成本偏高,但其作为有机化学原料,特别是三氟甲基化试剂还是具有很大的市场空间。将 CHF3直接作为三氟甲基化试剂还处于实验室初步探索阶段,其过程条件较为苛刻及成本较高,将其作为 CHF3处理路线还有待长时间探索。
[1]Han W F,Li Y,Tang H D,et al. Treatment of the potent greenhouse gas,CHF3-An overview[J]. Journal of Fluorine Chemistry,2012,140:7-16.
[2]杨虹,张剑波,冯金敏. 中国 HCFC-22 的消费与排放清单及预测[J]. 北京大学学报:自然科学版,2010,46(2):251-257.
[3]McCulloch A,Lindley A A. Global emissions of HFC-23 estimated to year 2015[J]. Atmos. Environ.,2007,41(7):1560-1566.
[4]Pantzali M N,Mouza A A,Paras S V. Pollutant emissions management in an existing plant:The CHF3case[J]. Chem. Eng.Technol.,2005,28(2):187-192.
[5]UNFCCC. AM0001:Incineration of HFC 23 waste streams[OL].2006-01-05 [2013-09-03]. http://cdm.unfccc.int/methodologies/index.html.
[6]Deam R T,Dayal A R,McAllister T,et al. Interconversion of chlorofluorocarbons in plasmas[J]. J. Chem. Soc.,Chem.Commun.,1995,3:347-348.
[7]Deam R T,Kearney T N,Ogilvy I M,et al. Material processing:EP,629138[P]. 1993-03-04.
[8]宋淑伟,史阿莹. 二氟一氯甲烷副产氯化氢的精制及三氟甲烷回收的方法与装置:中国,102101651 A[P]. 2011-10-02.
[9]Améduri B,Boutevin B,Kostov G. Fluoroelastomers:Synthesis,properties and applications[J]. Prog. Polym. Sci.,2001,26(1):105-187.
[10]Han W F,Kennedy E M,Mackie J C,et al. Synthesis of vinylidene fluoride via reaction of difluoromethane (HCFC-22)with methane[J]. Ind. Eng. Chem. Res.,2010,49(13):6010-6019.
[11]Tschuikow-Roux E,Marte J E. Thermal decomposition of fluoroform in a single-pulse shock tube[J]. J. Chem. Phys.,1965,42:2049-2056.
[12]Modica A P. Kinetics and equilibria of the difluorocarbene radical decomposition behind shock waves[J]. J. Chem. Phys.,1966,44(4):1585-1589.
[13]Politanskii S F,Shevchuk V U. Thermal conversions of fluoromethanes. II. Pyrolysis of di- and trifluoromethane[J]. Kinet.Catal.,1968,9(3):496-503.
[14]Tschuikow-Roux E. Thermal decomposition of fluoroform in a single-pulse shock tube. II. Pressure dependence of the rate[J]. J.Chem. Phys.,1965,42(6):3639 - 3642.
[15]Han W F,Kennedy E M,Kundu S K,et al. Experimental and chemical kinetic study of the pyrolysis of trifluoroethane and the reaction of trifluoromethane with methane[J]. Journal of Fluorine Chemistry,2010,131(7):751-760.
[16]Moon D J,Chung M J,Kim H,et al. Pyrolysis of trifluoromethane to produce hexafluoropropylene[J]. Ind. Eng. Chem. Res.,2002,41(12):2895-2902.
[17]Moon D J,Ahn B S. Pyrolysis of a mixture of trifluoromethane and tetrafluoroethylene to produce hexafluoropropylene[J]. Journal of Chemical Engineering of Japan,2004,37(2):318-325.
[18]劳 V N M,格尔布卢姆 P G,诺埃尔克 C J,等. 三氟甲烷(HFC23)的处理:中国,02825284.5[P]. 2005-04-06.
[19]Yu H,Kennedy E M,Ong W H,et al. Experimental and kinetic studies of gas-phase pyrolysis of n-C4F10[J]. Ind. Eng. Chem. Res.,2008,47(8):2579-2584.
[20]Han W F,Kennedy E M,Liu H Z,et al. Catalytic pyrolysis of CHF3over activated carbon and activated carbon supported potassium catalyst[J]. Journal of Fluorine Chemistry,2010,131(6):698-703.
[21]Sung D J,Moon D J,Moon S,et al. Catalytic pyrolysis of chlorodifluoromethane over metal fluoride catalysts to produce tetrafluoroethylene[J]. Applied Catalysis A—General,2005,292:130-137.
[22]Améduri B,Boutevin B. Update on fluoroelastomers:From perfluoroelastomers to fluorosilicones and fluorophosphazenes[J].Journal of Fluorine Chemistry,2005,126(2):221-229.
[23]Moore A L. Fluoroelastomers Handbook:the Definitive User’s Guide and Databook[M]. Norwich:William Andrew Publishing,2005:395.
[24]Améduri B. From vinylidene fluoride (VDF) to the applications of VDF-containing polymers and copolymers:Recent developments and future trends[J]. Chem. Rev.,2009,109(12):6632-6686.
[25]Li K,Kennedy E M,Dlugogorski B Z. Gas-phase reaction of halon 1301 (CBrF3) with methane[J]. Ind. Eng. Chem. Res.,1999,38:3345-3352.
[26]Li K,Kennedy E M,Dlugogorski B Z. Experimental and computational studies of the pyrolysis of CBrF3,and the reaction of CBrF3with CH4[J]. Chem. Eng. Sci.,2000,55(19):4067-4078.
[27]Tran R,Kennedy E M,Dlugogorski B Z. Gas-phase reaction of halon 1211 (CBrClF2) with methane[J]. Ind. Eng. Chem. Res.,2001,40(14):3139-3143.
[28]Yu H,Kennedy E M,Adesina A A,et al. A review of CFC and halon treatment technologies—The nature and role of catalysts[J].Catalysis Surveys from Asia,2006,10(1):40-54.
[29]Yu H,Kennedy E M,Mackie J C,et al. Simultaneous conversion of CHClF2and CH3Br to CH2CF2[J]. Chemosphere,2007,68(10):2003-2006.
[30]Uddin M A,Kennedy E M,Yu H,et al. Catalytic process for the conversion of halon 1211 (CBrClF2) to halon 1301 (CBrF3) and CFC 13 (CClF3)[J]. Ind. Eng. Chem. Res.,2003,42:6000-6006.
[31]Yu H,Mackie J C,Kennedy E M,et al. Experimental and quantum chemical study of the reaction CF2+ CH3CF2CH3—→CH2=CF2+ H:A key mechanism in the reaction between methane and fluorocarbons[J]. Ind. Eng. Chem. Res.,2006,45:3758-3762.
[32]Han W F,Kennedy E M,Mackie J C,et al. Conversion of a CFCs,HFCs and HCFCs waste mixture via reaction with methane[J].J. Hazard. Mater.,2010,184(1-3):696-703.
[33]Yu H,Kennedy E M,Mackie J C,et al. An experimental and kinetic modeling study of the reaction of CHF3with methane[J].Environmental Science & Technology,2006,40(18):5778-5785.
[34]Kiefer J H,Kumaran S S. Rate of CH4dissociation over 2800—4300 K:The low-pressure-limit rate constant[J]. J. Phys. Chem.,1993,97(2):414-420.
[35]Han W F,Kennedy E M,Mackie J C,et al. Mechanistic study of the reaction of CHF3with CH4[J]. Chemical Engineering Journal,2011,166(3):822-831.
[36]Han W F,Kennedy E M,Mackie J C,et al. Conversion of CHF3to CH2=CF2via reaction with CH4in the presence of CBrF3:An experimental and kinetic modelling study[J]. Journal of Hazardous Materials,2010,180(1-3):181-187.
[37]Han W F,Kennedy E M,Mackie J C,et al. Effect of methanol on the gas-phase reaction of trifluoromethane with methane[J]. Industrial& Engineering Chemistry Research,2010,49(18):8406-8414.
[38]Han W F,Yu H,Kennedy E M,et al. Conversion of CHF3to CH2=CF2via reaction with CH4and CaBr2[J]. Environmental Science & Technology,2008,42(15):5795-5799.
[39]马洋博,吕剑. 第三代ODS替代品FIC-1311[J]. 化工新型材料,2007,35(12):28-29.
[40]Nagasaki N,Morikuni Y,Kawada K,et al. Study on a novel catalytic reaction and its mechanism for CF3I synthesis[J]. Catalysis Today,2004. 88(3-4):121-126.
[41]周黎旸. 三氟碘甲烷应用进展[J]. 化工生产与技术,2009,16(4):5-6.
[42]马洋博,吕剑. 三氟碘甲烷合成和应用研究进展[J]. 有机氟化工,2007,14(4):8-10.
[43]马洋博,吕剑,张伟,等. 负载型固体碱催化合成三氟碘甲烷[J].化学试剂,2010,32(3):273-274.
[44]杨光成,石磊,毛燕超,等. 气相催化合成CF3I的催化剂载体研究[J]. 应用化工,2009,38(5):678-680.
[45]Yang G C,Jia X Q,Pan R M,et al. The disproportionation of CF2carbene in vapor-phase pyrolysis reaction over activated carbon and porous aluminum fluoride[J]. Journal of Molecular Catalysis A:Chemical,2009,309(1-2):184-188.
[46]Yang G C,Lei S,Pan R M,et al. Investigation of CF2carbene on the surface of activated charcoal in the synthesis of trifluoroiodomethane via vapor-phase catalytic reaction[J]. Journal of Fluorine Chemistry,2009,130(2):231-235.
[47]潘菲,施章杰. 过渡金属催化碳氢键三氟甲基化[J]. 化学学报,2012,70(16):1679-1681.
[48]Tomashenko O A,Grushin V V. Aromatic trifluoromethylation with metal complexes[J]. Chemical Reviews,2011,111(8):4475-4521.
[49]Zanardi A,Novikov M A,Martin E,et al. Direct cupration of fluoroform[J]. Journal of the American Chemical Society,2011,133(51):20901-20913.
[50]Novák P,Lishchynskyi A,Grushin V V. Fluoroform-derived CuCF3for low-cost,simple,efficient,and safe trifluoromethylation of aryl boronic acids in air[J]. Angewandte Chemie International Edition,2012,51(31):7767-7770.
[51]Prakash G K S,Jog P V,Batamack P T D,et al. Taming of fluoroform:Direct nucleophilic trifluoromethylation of Si,B,S,and C centers[J]. Science,2012,338(6112):1324-1327.
[52]Haufe G. Putting a greenhouse gas to work[J]. Science,2012,338(6112):1298.
猜你喜欢
建材发展导向(2021年14期)2021-08-23
有机氟工业(2020年2期)2020-07-04
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
中国煤层气(2019年2期)2019-08-27
环境与可持续发展(2017年2期)2017-04-06
合成化学(2015年10期)2016-01-17
有机氟工业(2014年3期)2014-06-05
火炸药学报(2014年1期)2014-03-20
影像科学与光化学(2014年5期)2014-03-11
中国医药科学(2013年16期)2013-12-20
